S-1: General form of registration statement for all companies including face-amount certificate companies
Published on
As filed with the Securities and Exchange Commission on September 20, 2023.
Registration No. 333-
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM
REGISTRATION STATEMENT UNDER THE SECURITIES ACT OF 1933
(Exact name of registrant as specified in its charter)
| 8731 | ||||
|
(State or other jurisdiction of incorporation or organization) |
(Primary Standard Industrial Classification Code Number) |
(I.R.S. Employer Identification Number) |
(Address, including zip code, and telephone number, including area code, of registrant’s principal executive offices)
Chief Executive Officer
(Name, address, including zip code, and telephone number, including area code, of agent for service)
Copies to:
|
Leslie Marlow, Esq. Hank Gracin, Esq. Patrick J. Egan, Esq. Blank Rome LLP 1271 Avenue of the Americas New York, New York 10020 Tel: (212) 885-5000 |
Ross David Carmel, Esq. Jeffrey P. Wofford, Esq. Carmel, Milazzo & Feil LLP 55 West 39th Street 4th Floor New York, New York 10018 (212) 658-0458 |
Approximate date of commencement of proposed sale to the public: As soon as practicable after the effective date of this registration statement.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933 check the following box: ☒
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer ☐ | Accelerated filer ☐ | |
| Smaller
reporting company |
||
| Emerging
growth company |
If
an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying
with any new or revised financial accounting standards provided pursuant to Section 7(a)(2)(B) of the Securities Act.
The Registrant hereby amends this Registration Statement on such date or dates as may be necessary to delay its effective date until the Registrant shall file a further amendment which specifically states that this Registration Statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933 or until the Registration Statement shall become effective on such date as the Commission, acting pursuant to said Section 8(a), may determine.
The information in this prospectus is not complete and may be changed. These securities may not be sold until the registration statement filed with the Securities and Exchange Commission is effective. This prospectus is not an offer to sell these securities, and it is not soliciting an offer to buy these securities in any jurisdiction where the offer or sale is not permitted.
Subject to Completion, dated September 20, 2023.
PRELIMINARY PROSPECTUS
3,086,419 Units
Each Unit Consisting of
One Share of Common Stock and
One Warrant to Purchase One Share of Common Stock,
(and the shares of Common Stock underlying such Warrants)
![]()
bioAffinity Technologies, Inc.
bioAffinity Technologies, Inc., a Delaware corporation headquartered in Texas (the “Company”), develops noninvasive, diagnostics to detect cancer and lung disease at early stage, and is researching targeted therapies to treat cancer at the cellular level.
This is the public offering (the “Offering”) of up to 3,086,419 units (each, a “Unit,” collectively, the “Units”). Each Unit consists of one share of our common stock, $0.007 par value per share (the “Common Stock”) and one warrant to purchase one share of Common Stock at an at an assumed offering price of $1.62 per Unit, which was the closing price of the Common Stock on September 13, 2023. The Units have no stand-alone rights and will not be certificated or issued as stand-alone securities. The shares of Common Stock and the Warrants underlying the Units are immediately separable and will be issued separately in this Offering. Each Warrant offered as part of this Offering will have an exercise price of $[●] per share (equal to 120% of the public offering price of each Unit sold in this offering), is immediately exercisable on the date of issuance and will expire five years from the date of issuance. The actual public offering price per Unit (the “Offering Price”) will be determined between the underwriters and us at the time of pricing, considering our historical performance and capital structure, prevailing market conditions, and overall assessment of our business.
Pursuant to the registration statement related to this prospectus, we are also registering the shares of Common Stock issuable upon exercise of Warrants.
Our Common Stock and our tradeable warrants issued in our initial public offering (the “Tradeable Warrants”) are listed on the Nasdaq Capital Market (“Nasdaq”) under the symbols “BIAF” and “BIAFW,” respectively. On September 13, 2023, the last reported sale price of our Common Stock was $1.62 per share and the last reported sale price of our Tradeable Warrants was $0.21. The actual Offering Price will be determined between us and WallachBeth Capital, LLC (“WallachBeth”) as the representative of the underwriters at the time of pricing, taking into consideration several factors as described in “Underwriting – Pricing of the Offer” and may be at a discount to the current market price. Therefore, the assumed public offering price used throughout this prospectus may not be indicative of the final offering price.
We are an “emerging growth company” and a “smaller reporting company” under applicable federal securities laws and will be subject to reduced public company reporting requirements.
Investing in our securities involves a high degree of risk. See the “Risk Factors” section beginning on page 14 of this prospectus for a discussion of the factors that you should consider before investing in our Common Stock.
Neither the Securities and Exchange Commission (the “SEC”) nor any state securities commission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
| Per Unit |
Total Assuming No Exercise of Over- Allotment Option |
Total With Full Exercise of Over- Allotment Option |
||||||||||
| Public Offering Price | $ | $ | $ | |||||||||
| Underwriting discount(1) | $ | $ | $ | |||||||||
| Proceeds, before expenses, to us(2) | $ | $ | $ | |||||||||
| (1) | We have agreed to issue, on the closing date of this Offering, a warrant to WallachBeth Capital, LLC, the representative of the underwriters (the “Representative”; such warrant, the “Representative’s Warrant”), to purchase an amount equal to two percent (2.0%) of the aggregate number of shares of Common Stock underlying the Units sold by us in this Offering. The Representative’s Warrant is exercisable for a period of five years, commencing on the date that is 180 days after the commencement date of sales of the Units in this Offering and expiring on the five year anniversary of the effective date of the registration statement of the Offering. Please read the section titled “Underwriting” for a description of all underwriting compensation payable by us in connection with this Offering. | |
| (2) | The amount of Offering proceeds to us presented in this table does not give effect to any exercise of the Warrant we will issue to the Representative, as described herein. |
We have granted the underwriters a 45-day option from the date of this prospectus to purchase up to a total of an additional 462,962 shares of Common Stock at $[●] per share (the Offering Price less $0.01), and/or 462,962 Warrants at $0.01 per Warrant, or any combination of additional shares of Common Stock and Warrants representing, in the aggregate, up to 15% of the number of Units sold in this Offering (the “Over-Allotment Option”), in all cases less the underwriting discount.
The underwriters expect to deliver the Units to purchasers on or about [●], 2023 through the book-entry facilities of The Depository Trust Company.
WallachBeth Capital, LLC
Craft Capital Management LLC
The date of this prospectus is September [●], 2023.
bioAffinity Technologies, Inc.
TABLE OF CONTENTS
MARKET, INDUSTRY, AND OTHER DATA
About this Prospectus
You should rely only on the information contained in this prospectus prepared by us or on our behalf or to which we have referred you. We have not, and the underwriters have not, authorized any other person to provide you with information different from that contained in this prospectus. If anyone provides you with different or inconsistent information, you should not rely on it. Neither we nor the underwriter take responsibility for and can provide no assurance as to the reliability of, any other information that others may give you. We are not, and the underwriters are not, making an offer to sell the securities described herein in any jurisdiction where an offer or sale is not permitted. The information in this prospectus, or any free writing prospectus, is accurate only as of the date of this prospectus, regardless of the time of delivery of this prospectus or any sale of our Units. Our business, financial condition, results of operations, and prospects may have changed since that date.
This prospectus contains forward-looking statements that are subject to a number of risks and uncertainties, many of which are beyond our control. Please read “Risk Factors” and “Cautionary Note Regarding Forward-Looking Statements.”
Unless the context otherwise requires, the information in this prospectus (other than in the historical financial statements) assumes that the underwriters will not exercise their option to purchase additional shares of our common stock, $0.007 par value per share (the “Common Stock”) or additional warrants to purchase shares of Common Stock (the “Warrants”).
For investors outside of the United States: We are not making an offer of any securities in any jurisdiction in which such offer is unlawful. Neither we nor any of the underwriters have done anything that would permit this Offering or possession or distribution of this prospectus or any free writing prospectus we may provide to you in connection with this Offering in any jurisdiction where action for that purpose is required, other than in the United States. Persons outside of the United States who come into possession of this prospectus and any free writing prospectus must inform themselves about and observe any restrictions relating to this Offering and the distribution of this prospectus outside of the United States. See “Underwriting—Selling Restrictions” on page 110.
Industry and Market Data
This prospectus includes estimates regarding market and industry data. Unless otherwise indicated, information concerning our industry and the markets in which we operate, including our general expectations, market position, market opportunity, and market size, are based on our management’s knowledge and experience in the markets in which we operate, together with currently available information obtained from various third-party sources, including publicly available information, industry reports and publications, surveys, our customers, trade and business organizations, and other contacts in the markets in which we operate. Although we believe these third-party sources are reliable as of their respective dates, neither we nor the underwriters have independently verified the accuracy or completeness of this information. Some data is also based on our good faith estimates. The industry in which we operate is subject to a high degree of uncertainty and risk due to a variety of factors, including those described in the section entitled “Risk Factors.” These and other factors could cause results to differ materially from those expressed in these publications.
Trademarks and Trade Names
We own or have rights to various trademarks, service marks, and trade names that we use in connection with the operation of our business. This prospectus may also contain trademarks, service marks, and trade names of third parties, which are the property of their respective owners. Our use or display of third parties’ trademarks, service marks, trade names, or products in this prospectus is not intended to, and does not imply a relationship with or endorsement or sponsorship by us. Solely for convenience, the trademarks, service marks, and trade names referred to in this prospectus may appear without the ®, TM or SM symbols, but such references are not intended to indicate, in any way, that we will not assert, to the fullest extent under applicable law, our rights or the right of the applicable licensor to these trademarks, service marks, and trade names.
| 1 |
PROSPECTUS SUMMARY
This summary provides an overview of information appearing elsewhere in this prospectus and highlights the key aspects of this Offering. This summary does not contain all of the information you should consider prior to investing in our Common Stock or Warrants. You should read this entire prospectus carefully, including the sections titled “Risk Factors” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our consolidated financial statements and related notes appearing at the end of this prospectus, before making any investment decision. Our fiscal year ends on December 31. Unless the context otherwise requires, references to “bioAffinity,” the “Company,” “we,” “us,” and “our” in this prospectus refer to bioAffinity Technologies, Inc. and our consolidated subsidiaries.
Overview
bioAffinity Technologies, Inc. (the “Company,” “we,” or “our”) develops noninvasive diagnostics to detect early-stage lung cancer and other diseases of the lung. We are developing our platform technologies so that, in the future, they could result in broad-spectrum cancer treatments. We develop proprietary noninvasive diagnostic tests using technology that preferentially targets cancer cells and cell populations indicative of a diseased state.
We were formed as a Delaware corporation on March 26, 2014. On June 15, 2016, we formed OncoSelect® Therapeutics, LLC (“OncoSelect®”), a Delaware limited liability company and our wholly owned subsidiary. On August 14, 2023, we formed Precision Pathology Laboratory Services, LLC (“PPLS”), a Texas limited liability company and our wholly owned subsidiary. Research and optimization of our platform technologies are conducted in our laboratories at The University of Texas at San Antonio.
Our first diagnostic test, CyPath® Lung, addresses the need for noninvasive detection of early-stage lung cancer. Lung cancer is the leading cause of cancer-related deaths. Physicians will be able to order CyPath® Lung to assist in their assessment of patients who are at high risk for lung cancer. The CyPath® Lung test enables physicians to more confidently identify patients who will likely benefit from timely intervention and more invasive follow-up procedures and is another tool to help distinguish them from patients who are likely without lung cancer and should continue annual screening. CyPath® Lung has the potential to increase overall diagnostic accuracy of lung cancer, which could lead to increased survival, fewer unnecessary invasive procedures, reduced patient anxiety, and lower medical costs.
Through our wholly owned subsidiary, OncoSelect®, our research has led to discoveries of novel potential cancer therapeutics that specifically and selectively target cancer cells that have been grown in petri dishes.
Recent Developments
On September 18, 2023, PPLS, our wholly owned subsidiary, consummated the acquisition (the “Acquisition”) of a clinical anatomic and clinical pathology laboratory and related services business in San Antonio, Texas (the “ Laboratory Assets”) pursuant to the terms of an Asset Purchase Agreement (the “Asset Purchase Agreement”) dated September 18, 2023 that it entered into with Village Oaks Pathology Services, P.A., a Texas professional association d/b/a Precision Pathology Services (“Village Oaks”) and Dr. Roby P. Joyce, M.D. As a result of the Acquisition, the clinical pathology laboratory is owned by PPLS. Dr. Joyce was the Medical Director and Laboratory Director of the clinical pathology laboratory prior to the Acquisition and he continues to serve as Medical Director and Laboratory Director after the Acquisition. PPLS is accredited by the College of American Pathologists (“CAP”) and certified under the Clinical Laboratory Improvement Amendments of 1988 (“CLIA”). Founded in 2007 by Dr. Roby Joyce, Village Oaks has provided pathology services to physicians practicing in a variety of outpatient settings. Since September 2021, Village Oaks, under the trade name Precision Pathology Services, has offered for sale CyPath® Lung as a laboratory developed test (“LDT”) for the detection of early-stage lung cancer. In addition to CyPath® Lung, PPLS intends to continue to offer a range of laboratory services including respiratory testing for SARS-CoV-2 and influenza, anatomical pathology, morphological stains, histological services, DNA extractions, STI testing and women’s and men’s health testing.
Pursuant to the terms of the Asset Purchase Agreement, PPLS acquired the Laboratory Assets, which included all of the assets owned by Village Oaks other than medical assets, which are assets Village Oaks used in connection with its management and operation of a clinical pathology laboratory, now owned by PPLS, and related services business and assumed certain liabilities and obligations. Pursuant to the terms of the Asset Purchase Agreement Village Oaks received $3,500,000 in consideration for the assets to be purchased by PPLS, of which $1,000,000 was paid by the issuance of 564,972 shares of our restricted Common Stock to a trust controlled by Dr. Joyce (the “Joyce Trust”), which share number was determined by dividing $1,000,000 by $1.77, the average of the trading day closing prices for the 30 days prior to September 15, 2023, rounded to the nearest whole share.
The Asset Purchase Agreement contains customary representations, warranties and covenants made by PPLS and Village Oaks and consummation of the transaction was subject to customary closing conditions, including, among other things, entry into the other ancillary agreements described below. Subject to certain customary limitations, Village Oaks agreed to indemnify PPLS, its successors and assigns, and each of their affiliates, and PPLS’ officers, directors, employees and other authorized agents against certain losses related to, among other things, breaches of Village Oaks’ representations, warranties, covenants and agreements as well as any excluded liabilities and excluded assets described therein. Subject to certain customary limitations, PPLS also agreed to indemnify Village Oaks, its successors and assigns, and each of their affiliates, and Village Oaks’ officers, directors, employees and other authorized agents against certain losses related to, among other things, breaches of PPLS’ representations, warranties, covenants and agreements as well as any assumed liabilities.
Pursuant to the Asset Purchase Agreement, PPLS assumed all liabilities and obligations and obtained any and all rights, title and interest of Village Oaks in and to (i) all leases for equipment and personal property related to the Laboratory Assets (the “Assumed Leases”), pursuant to an Assumption Agreement by and between Village Oaks and PPLS (the “Assumption Agreement”) and, (ii) certain other contracts related to the Laboratory Assets, including the license to develop, manufacture, use, market and sell CyPath® Lung (the “Assumed Contracts”) pursuant to the Assumption Agreement; (iii) all accounts payable of Village Oaks as of September 18, 2023 that were incurred in the ordinary course of business consistent with past custom and practice; and (iv) the lease of the premises used in connection with operation of the CLIA-certified and CAP-accredited clinical pathology laboratory, pursuant to an Assignment and Assumption of Lease by and between Village Oaks and PPLS (the “Assignment of Lease”), which Assignment of Lease was consented to by the landlord of the leased premises. The monthly rent is currently $10,143.83 per month and the term of the Lease is five years.
| 2 |
In connection with the Asset Purchase Agreement, PPLS entered into a Management Services Agreement with Village Oaks (the “Management Services Agreement”) pursuant to which PPLS will provide comprehensive management and administrative services to Village Oaks in connection with the operation of Village Oaks’ professional cytopathology, histopathology, clinical and anatomic pathology interpretation medical services practice. PPLS will provide space, equipment, administrative, management and clinical personnel, billing and collection, and related management services to Village Oaks in exchange for a management fee of 70% of the net revenues received by Village Oaks from the provision of the medical services. The Management Services Agreement has an initial term of 20 years and provides that upon expiration of the initial term, it will be automatically extended for two additional successive terms of five years each, unless either party delivers written notice of its intention not to extend the term of the agreement not less than 90 days prior to the expiration of the preceding term. The Management Services Agreement also provides that until the fifth anniversary of its effective date, Village Oaks will not, without the prior written approval of PPLS own, operate or have any financial interest in any other person or entity that operates an independent laboratory or an enterprise within the United States that provides or promotes management or administrative services or any product or services substantially similar to those provided by PPLS.
In connection with the Asset Purchase Agreement, PPLS entered into a Succession Agreement with Village Oaks and Dr. Joyce (the “Succession Agreement”), pursuant to which Dr. Joyce, as holder of 100% of the issued and outstanding stock of Village Oaks, and Village Oaks are restricted from disposing of their equity interests in Village Oaks, subject to certain exceptions, without the prior written consent of us and Village Oaks. The Succession Agreement further provides that the entire equity interest held by Dr. Joyce in Village Oaks will be automatically assigned and transferred to a successor who meets the Eligibility Requirements of a Designated Physician, as such terms are defined and described in the Succession Agreement, in the event of, among other things, the death, disability, retirement, or a court’s determination of incompetence of Dr. Joyce, as well as Dr. Joyce’s failure to satisfy the eligibility requirements of a Designated Physician, exclusion or disqualification from participation in the Medicare program, conviction of a felony or crime or moral turpitude, bankruptcy filing, or material breach of the Succession Agreement. In the event of the automatic transfer of Dr. Joyce’s equity interests in Village Oaks as provided in the Succession Agreement, such agreement provides that the board of directors of Village Oaks shall nominate a group of three candidates as the Designated Physician who satisfy the Eligibility Requirements. In the event the Company desires not to select any of such candidates, the Company shall select and appoint a successor Designated Physician from any other physicians that satisfy the Eligibility Requirements. Subject in all cases to the Management Services Agreement, Dr. Joyce shall not cause any voluntary interruption of the conduct of Village Oaks’ business and operations, and shall use commercially reasonable efforts to preserve (or assist us in preserving) all rights, privileges and franchises held by Village Oaks, including the maintenance of all contracts, copyrights, trademarks, licenses and registrations.
In connection with the Asset Purchase Agreement, PPLS entered into a Professional Services Agreement with Village Oaks (the “Professional Services Agreement”) pursuant to which Village Oaks will provide pathology interpretation services as requested on behalf of PPLS based on the professional fees approved for the CPT code for the services provided under the Medicare Physician Fee Schedule in the locality where the test is performed. The Professional Services Agreement has an initial term of 20 years and provides that upon expiration of the initial term, it will be automatically extended for successive terms of twelve months each, unless either party delivers written notice of its intention not to extend the term of the agreement not less than 30 days prior to the expiration of the preceding term.
In connection with the Asset Purchase Agreement, we entered into an Executive Employment Agreement with Dr. Joyce (the “Joyce Employment Agreement”), for a term of three years, pursuant to which he serves as the Medical Director and Laboratory Director of PPLS, at a base salary of $333,333.34 per year. Pursuant to the Joyce Employment Agreement, Dr. Joyce was also appointed to serve on our Board of Directors. Dr. Joyce will be eligible to participate in or receive benefits under our benefit plans generally made available to executives of similar status and responsibilities and will be provided use of a company car. In the event the Joyce Employment Agreement is terminated for any reason, including by Dr. Joyce upon 60 days’ notice, by us for cause or by reason of Dr. Joyce’s death, Dr. Joyce (or his estate, as applicable) will receive his base salary for the remainder of the three-year employment term. However, the Joyce Employment Agreement provides that if Dr. Joyce breaches any of the restrictive covenants set forth in the Joyce Employment Agreement, including a covenant not to compete during his term of employment and a covenant not to knowingly disclose confidential information, such breach will be grounds for the immediate termination of Dr. Joyce and will result in the forfeiture of all compensation and benefits otherwise due to Dr. Joyce.
One of the Assumed Leases is Equipment Usage Attachment, dated effective as of August 9, 2019, by and between Gen-Probe Sales & Service, Inc., together with its subsidiaries and affiliates (“Hologic”) and Village Oaks, as amended by that certain Amendment No. 1 to Equipment Usage Attachment dated November 2, 2020, as further amended by that certain Amendment No. 2 to Equipment Usage Attachment dated November 2, 2020, and as further amended by that certain Amendment No. 3 to Equipment Usage Attachment dated December 21, 2022 (the “Hologic Equipment Lease”), pursuant to which PPLS leases reagent equipment from Hologic and is required to purchase a minimum number of specified testing kits each year. The total monthly minimum purchase commitment PPLS is required to pay Hologic, inclusive of the lease of the reagent equipment, is $16,914 per month. The term of the Hologic Equipment Lease currently expires on December 20, 2027.
Another of the Assumed Leases is the Master Agreement, dated as of January 29, 2015, by and between Leica Microsystems, Inc. (“Leica”) and Village Oaks, as amended by Amendment No. 1 to the Master Agreement, dated on or about April 4, 2018, as further amended by that certain Amendment No. 2 to Master Agreement, dated March 23, 2021 (the “Leica Equipment Lease”), pursuant to which PPLS leases reagent equipment from Leica and is required to purchase a minimum number of specified testing kits. The total monthly minimum purchase commitment PPLS is required to pay to Leica, inclusive of the lease of the reagent equipment, is $19,790 per month. The term of the Leica Equipment Lease currently expires on March 23, 2026.
| 3 |
One of the Assumed Contracts is a Strategic Relationship License Agreement, dated December 1, 2022, by and between Pathology Watch, Inc. (“Pathology Watch”) and Village Oaks (the “License Agreement”). Pursuant to the License Agreement, Pathology Watch granted a license to its digital imaging cloud-based pathology platform to facilitate remote interpretation and billing of pathology specimens by qualified professionals to PPLS for a monthly fee of $25,000. In connection with the License Agreement, Pathology Watch also provides certain support services and marketing vendor services to PPLS for the monthly fee of $38,000, for a total monthly fee paid by PPLS to Precision Watch of $63,000. The License Agreement is for an initial term of twelve months, unless terminated by either party upon 90 days’ notice, and provides that upon expiration of the initial term (or any renewal term), it will be automatically extended for successive twelve month terms, unless either party notifies the other party of its intention not to renew the License Agreement not less than 90 days prior to the expiration of the current term.
In connection with the Asset Purchase Agreement, Dr. Joyce, on behalf of Village Oaks, executed a Bill of Sale (the “Bill of Sale”), pursuant to which all rights, title, and interest of Village Oaks in and to the permits listed on Exhibit A attached thereto, inclusive of the CLIA-certificate and CAP-accreditation, notwithstanding the transfer of the CLIA certificate by operation of law to PPLS upon consummation of the Acquisition, were confirmed to have been transferred and assigned to PPLS.
Benefits of the Acquisition
Since 2007, Village Oaks, d/b/a Precision Pathology Services, has provided pathology services to physicians practicing in a variety of outpatient settings. Since September 2021, Village Oaks has offered CyPath® Lung as an LDT for the detection of early-stage lung cancer. The Acquisition of the Laboratory Assets supports and enhances our commercial strategy by integrating every aspect of CyPath® Lung, from manufacturing to sales and marketing, and to pathology services and reporting results back to physicians. The Acquisition results in consolidating the royalties to bioAffinity from sale of CyPath® Lung with the revenues derived by PPLS’ sale of the test. The Acquisition provides us with control over essential aspects of the CyPath® Lung test and an opportunity to build scale and efficiency as we expand commercialization. In addition, we anticipate the Acquisition can support our pivotal trial by providing the equipment, experienced laboratory personnel and administrative support including support for our planned United States (“U.S.”) Food and Drug Administration (the “FDA”) clinical study. Ownership of the Laboratory Assets also provides the clinical services required for us to develop future LDTs that expand our flow cytometry platform, including development of current research into a test for Chronic Obstructive Pulmonary Disease (“COPD”). Village Oaks’ operation of a CLIA-certified and CAP-accredited clinical pathology laboratory has been established over the past 16 years of service. The founder of Village Oaks, Dr. Joyce, will continue as PPLS’ Medical Director and Laboratory Director, thus providing continuity in professional relationships and services. PPLS intends to continue to offer a range of laboratory services in addition to CyPath® Lung, including respiratory testing for SARS-CoV-2 and influenza, anatomical pathology, morphological stains, histological services, DNA extractions, STI testing and women’s and men’s health testing.
Amendment to Warrants
On September 17, 2023, Mr. Girgenti, the Cranye Girgenti Testamentary Trust, Gary Rubin, The Harvey Sandler Revocable Trust, a trust of which Mr. Rubin is a co-trustee, Ms. Zannes and Dr. Joyce consented to an amendment of the terms of the outstanding warrants that they own. Such warrants include warrants (i) tradeable warrants (the “Tradeable Warrants”) to purchase 98,198, 39,182, and 39,182 shares of Common Stock owned by Mr. Girgenti, The Harvey Sandler Revocable Trust, and Ms. Zannes, respectively); (ii)non-tradeable warrants (the “Non-Tradeable Warrants”) to purchase 102,286, 40,813, and 40,813 shares of Common Stock owned by Mr. Girgenti, The Harvey Sandler Revocable Trust, and Ms. Zannes, respectively; and (iii) other outstanding warrants (the “Pre-IPO Warrants”) to purchase 469,063, 8,332, 571,373, 23,571, 17,137, and 14,285 shares of Common Stock owned by Mr. Girgenti, the Cranye Girgenti Testamentary Trust, Mr. Rubin, The Harvey Sandler Revocable Trust, Ms. Zannes and Dr. Joyce, respectively. The warrant amendment (the “Warrant Amendment”) provides that such warrants will not be exercisable until the date that we file a certificate of amendment to our certificate of incorporation with the State of Delaware which increases the number of shares of our authorized Common Stock to allow for sufficient authorized and unissued shares of Common Stock for the full exercise of all of the outstanding Pre-IPO Warrants, Tradeable Warrants and Non-Tradeable Warrants of the Company and the issuance of all of the shares of Common Stock underlying such warrants.
Financial
To date, we have devoted a substantial portion of our efforts and financial resources to the development of our first diagnostic test, CyPath® Lung. Since our inception in 2014, we have funded our operations principally through public and private sales of our equity or debt securities. On September 6, 2022, we completed our initial public offering of our securities pursuant to which we raised gross proceeds of $7.9 million. As of September 18, 2023, investors participating in the initial public offering exercised a total of 725,576 Tradeable Warrants at a price of $7.35 per share and 310,910 Non-Tradable Warrants at a price of $7.656 per share. Combined with our underwritten public offering, we received an aggregate of approximately $15.6 million as of September 28, 2022. We believe that our available cash together with the proceeds of this Offering will be sufficient to fund our planned operations for at least 12 months following the date of this prospectus.
In the second quarter of 2022, we started to recognize revenue from sales of the CyPath® Lung test by Village Oaks, a CAP-accredited and CLIA-certified clinical pathology laboratory to which we had previously granted a license to develop CyPath® Lung for commercialization and to manufacture, use, market and sell CyPath® Lung as an LDT prior to the Acquisition, which license was assigned to and assumed by PPLS, our wholly owned subsidiary, in connection with the Acquisition. We have never been profitable, and as of June 30, 2023, we had total working capital of $7.9 million and an accumulated deficit of approximately $39.9 million. As of June 30, 2023, we had cash and cash equivalents of $8.3 million. We expect to continue to incur significant operating losses for the foreseeable future as we continue the development of our diagnostic tests and therapeutic products and advance our diagnostic tests through clinical trials.
We anticipate raising additional cash needed through the private or public sales of equity or debt securities, collaborative arrangements, or a combination thereof, to continue to fund our operations and develop our products. There is no assurance that any such collaborative arrangement will be entered into or that financing will be available to us when needed in order to allow us to continue our operations, or if available, on terms acceptable to us. If we do not raise sufficient funds in a timely manner, we may be forced to curtail operations, delay our clinical trials, cease operations altogether, or file for bankruptcy.
Our First Diagnostic Test - CyPath® Lung
Lung cancer remains the most commonly diagnosed cancer and the leading cause of cancer-related deaths worldwide. Globally, there were an estimated 2.1 million lung cancer cases and 1.8 million lung cancer deaths in 2018, as reported by the World Health Organization in its 2018 Cancer Fact Sheet. According to the American Lung Association (the “ALA”), screening for individuals at high risk for lung cancer has the potential to improve lung cancer survival rates by finding disease at an earlier stage when it is more likely to be curable. A study published in the New England Journal of Medicine entitled “Survival of patients with stage I lung cancer detected on CT screening” dated October 26, 2006 reported that the survival rate of individuals with Stage I lung cancer who underwent surgical resection within one month after diagnosis had a ten-year survival rate of 92%, as compared to the overall five-year survival rate of 25%. Unfortunately, most lung cancer is detected in late stages. The results of a large national clinical trial that was reported in the New England Journal of Medicine in an article dated August 4, 2011, entitled “Reduced Lung-Cancer Mortality with Low-Dose Computed Tomographic Screening” showed that screening for lung cancer using low-dose computed tomography (“LDCT”) resulted in a reduction of the mortality rate by 20% as compared to screening by x-ray if LDCT screening is used by patients at high risk for lung cancer on an annual basis. Therefore LDCT scans are recommended for screening of an estimated 14 million Americans who are at high risk for lung cancer. If half of these high-risk individuals were screened, over 12,000 lung cancer deaths could be prevented, according to the ALA. However, the New England Journal of Medicine article also reported that LDCT was shown to have a low positive predictive rate of less than 4%. This means that for every 100 people who receive a positive result from LDCT screening and are suspected of having lung cancer, only four of those patients truly have the disease. A reliable, noninvasive and cost-effective diagnostic test can increase diagnosis of early-stage lung cancer while lowering the number of unnecessary and invasive procedures for patients with a false positive result from LDCT screening. (False positive means a person who does not have lung cancer but receives a positive result, in this case from LDCT screening.)
| 4 |
CyPath® Lung is a test for early-stage lung cancer that is designed to meet the need for greater diagnostic certainty. Based on our internal analysis, its use in conjunction with LDCT is predicted to improve the positive predictive value (the probability that patients with a positive LDCT scan truly have the disease) by a factor of five. Our analysis concludes that improving the positive predictive value of LDCT with the use of CyPath® Lung has the potential to subject fewer patients to the stresses of misdiagnosis or unnecessary diagnostic procedures such as biopsies, while also reducing healthcare costs.
CyPath® Lung uses flow cytometry technology to detect and analyze cell populations in a person’s sputum, or phlegm, to find characteristics indicative of lung cancer, including cancer and/or cancer-related cells that have shed from a lung tumor. The flow cytometer is a well-established instrument used in many commercial laboratories that records properties of labeled and unlabeled single cells. Sputum is an excellent sample for analysis because it is in direct contact with any malignancy in the lungs and can thus provide a snapshot of the tumor itself, its microenvironment, and its area of field cancerization. While studies have shown that expert cytological analysis of sputum can detect cancerous and pre-malignant cells, the level of scrutiny required for the analysis is not feasible in the laboratory routine, according to an October 22, 2009 article, “Premalignant and malignant cells in sputum from lung cancer patients,” published in Cancer Cytopathology. The process of looking at microscopy slides is an extremely laborious approach and demands years of expertise. CyPath® Lung uses flow cytometry and automated data analysis developed by artificial intelligence (“AI”) that allows for an entire sample of sputum to be examined for cost-effective, large-scale screening or diagnosis.
In particular, CyPath® Lung uses a synthetic porphyrin called meso-tetra (4-carboxyphenyl) porphyrin (“TCPP”). Porphyrins are biological pigments that, when exposed to ultraviolet light at certain wavelengths, can result in the cell fluorescing a red or purplish color that can be detected under a microscope or by flow cytometry, according to an article entitled “Laboratory Diagnosis of Porphyria,” published in Diagnostics (Basel) on July 26, 2021. Porphyrins can be man-made, like TCPP, or they can be naturally occurring, like heme that is responsible for the red color in red blood cells. Cancer cells are known to take up certain porphyrins in higher amounts than non-cancer cells, and the high affinity for cancer cells displayed by TCPP makes it an excellent bio-label for cancer, according to an article published in Progress in Clinical and Biological Research in 1984 entitled, “A comparative study of 28 porphyrins and their abilities to localize in mammary mouse carcinoma: uroporphyrin I superior to hematoporphyrin derivative.” As used in CyPath® Lung, the proportion of cells with high TCPP fluorescence intensity in a patient’s sputum sample is a significant predictor of lung cancer. We hold multiple patents protecting our use of TCPP for the diagnosis, monitoring, and treatment of cancer. In addition, we have multiple domestic and foreign patent applications to protect the use of flow cytometry and our AI-developed automated analysis platform in the detection of lung cancer and other lung diseases using sputum as a sample.
We developed an algorithm as part of a test validation trial that used machine learning to distinguish samples from high-risk patients who had lung cancer from those who are cancer-free. Village Oaks developed CyPath® Lung for sale as an LDT in accordance with the standards of the CAP and the regulations and guidance of the CLIA program, which is administered by the Centers for Medicare and Medicaid Services (“CMS”). Our test can analyze an average sputum sample containing about 14 million cells in approximately 30 minutes using integrated software for high-throughput, user-friendly standardized analysis of flow cytometric sample data. A physician’s report is generated within minutes after data acquisition. The test can be put into routine lab use without requiring expert evaluation of samples or being subject to operator bias. Our approach allows the entire sputum sample to be rapidly analyzed. The numerical analysis developed with machine learning captures complex interactions between lung cancer, the microenvironment, and areas of field cancerization that would be difficult if not impossible for individuals to predict or detect reliably by eye. For example, during test development, we discovered that viability staining density suggests a link with apoptosis, or cell death, that is linked to many cancers, including lung cancer. Our model also suggests that specific markers of immune cell populations may be informative as to the presence of cancer in the lung. These findings are the result of our machine learning approach to automated analysis.
The CyPath® Lung diagnostic process uses sputum that is obtained noninvasively in the privacy of a patient’s home. Physicians can order the test for patients they suspect have lung cancer or patients with a positive LDCT screening result. A patient collects his or her sample using a hand-held, noninvasive assist device, ICU Medical’s acapella® Choice Blue, that acts to break up mucus in the lungs and help a person cough up sputum from the lung into a collection cup. The acapella® Choice Blue has been 510(k) cleared by the FDA as a positive expiratory pressure device to help mobilize lung secretions in people with certain lung conditions. The sputum sample is shipped overnight to a clinical pathology laboratory that is accredited by the CAP and certified under the CLIA program, and processed with CyPath® Lung that includes antibodies that distinguish different cell types and the synthetic porphyrin TCPP that identifies cancer cells and/or cancer-associated cells. Proprietary automated analysis software developed by bioAffinity Technologies analyzes sample data in minutes, resulting in a patient report provided to the physician who orders the test. CyPath® Lung can be used by physicians to find early-stage lung cancer in their patients who have undergone lung cancer screening.
We conducted a 150-patient test validation trial of people at high risk for lung cancer including patients with the disease (N=28) and those cancer-free (N=122) that resulted in CyPath® Lung’s overall 88% specificity, meaning the ability to correctly identify a person without cancer, and 82% sensitivity, meaning the ability to correctly identify cancer in a person with the disease. For the subset of patients in this trial who had lung nodules smaller than 20 millimeters (“mm”) or no nodules at all, this trial resulted in 92% sensitivity and 87% specificity. In this subset of 132 individuals with small nodules, 119 patients were cancer-free and 13 had confirmed lung cancer. The detection of small lung nodules in people who have early-stage cancer can increase lung cancer survival.
In this 19-month test validation trial, participants provided a sputum sample and were released from the study after a physician either confirmed the individual was cancer-free by examination of CT imaging or confirmed the presence of lung cancer by biopsy. Flow cytometry and patient data used in the analysis produced results that included (1) the proportion of cells with a high ratio of high TCPP fluorescence intensity over cell size; (2) the proportion of cells with an intermediate ratio of fluorescence intensity caused by the viability dye (FVS510) over cell size; (3) the proportion of cells that were CD206 negative but positive for one or more of the following markers: CD66b (granulocytes), CD3 (T cells), and CD19 (B cells); and (4) patient age.
The CyPath® technology is based on research originating at Los Alamos National Laboratory in collaboration with St. Mary’s Hospital (Colorado) in which cancer samples were differentiated from non-cancer samples with 100% accuracy. This early research was conducted with sputum from 12 uranium miners. Microscope slides were made of the sputum samples labeled with the active ingredient of CyPath®, the synthetic and fluorescent porphyrin TCPP. The Los Alamos research study of 12 uranium miners included eight men with cancer and four healthy individuals. Researchers were blinded to the sample origin and looked for the presence of highly fluorescent cells indicating uptake of TCPP and the presence of lung cancer. The length of the study and specific follow-up was not reported, but researchers did report that one patient entering the study as a healthy subject was correctly diagnosed with cancer by the test.
| 5 |
We conducted market research with pulmonologists, oncologists, cardiothoracic surgeons, radiologists, and internists engaged in the diagnosis and treatment of lung cancer to help assess these stakeholders’ reactions to the new diagnostic test. Research revealed a strong interest in CyPath® Lung, driven by the high level of unmet clinical need for noninvasive diagnostics. A survey conducted with 240 pulmonologists and internists, the primary audience for the test, showed that 96% would use CyPath® Lung if it were available today as an adjunct to LDCT screening and diagnosis. Physicians responded favorably to a noninvasive diagnostic technology that gives them more confidence in their decision to proceed with more aggressive follow-up procedures if the test comes back positive. If test results are negative, physicians could rule out lung cancer, thus reducing the number of costly invasive procedures that result from the LDCT false-positive rate.
Physicians can order the CyPath® Lung laboratory test for use by people at high risk for lung cancer who are recommended for annual screening by LDCT. While LDCT is shown to lower the mortality rate of lung cancer by at least 20% as compared to x-ray screening, the LDCT screening method has a low positive predictive value that can result in many people undergoing unnecessary invasive diagnostic procedures to confirm or rule out the presence of lung cancer. A physician who orders a CyPath® Lung test can have greater confidence in determining the next steps in patient care. Noninvasive sample collection and the test’s three-day turnaround in providing patient results after sample receipt make CyPath® Lung well suited for both sophisticated and less developed markets. On June 6, 2023, the American Medical Association (“AMA”) approved a Current Procedural Terminology (“CPT”) Proprietary Laboratory Analysis (“PLA”) code specifically for use with CyPath® Lung, which code was publicly released on June 30, 2023. The new CPT code will be effective October 1, 2023. Prior to and in the interim until the new code is effective, CyPath® Lung is reported with a non-specific CPT code, for which payment is determined by the payer on a case-by-case basis. Payment for the new PLA code was discussed on July 19, 2023, by the Medicare Advisory Panel on Clinical Diagnostic Laboratory Tests. The CMS preliminary determination will be released in September 2023 followed by a 30-day comment period. A 2024 payment determination, effective January 1, 2024 will be made by CMS in November 2023. There is an opportunity for reconsideration in next year’s payment cycle.
We have an agreement with GO2 Partners to produce patient collection kits and to provide warehousing and distribution services for sending out the kits. Laboratory reagents, supplies and equipment are commercially available through multiple vendors. Sample processing, labeling, and data collection can be accomplished by a laboratory technician skilled in general laboratory techniques. Data analysis leading to a physician’s report is done by automated analysis software fully integrated into the test.
To our knowledge, CyPath® Lung is the first cancer diagnostic that combines automated flow cytometric analysis to predict the presence of lung cancer from sputum samples.
OncoSelect® Therapeutics Research
OncoSelect® Therapeutics, LLC, a Delaware limited liability company and our wholly-owned subsidiary, is a preclinical-stage biopharmaceutical discovery company with a focus on therapeutics that deliver cytotoxic (cell-killing) effects on a broad selection of human cancers from diverse tissues while having little or no effect on normal cells.
Unlike many of our industry competitors, OncoSelect® does not pursue therapies that depend on specific mutations, biomarkers, or other genetic or epigenetic abnormalities for their effect. We pursue research based on our own scientific discoveries demonstrating that inhibition of the expression of two specific cell membrane proteins results in the selective killing of various cancer cell types grown in the laboratory with little or no effect on normal (non-cancerous) cells.
Our scientific discoveries stemmed from research we conducted to better understand the mechanism by which TCPP, the synthetic porphyrin used in CyPath® Lung, selectively enters cancer cells. We have established several specific areas of therapeutic research that have evolved from our TCPP experiments.
OncoSelect® therapies offer the possibility of broad applications in cancer treatment. OncoSelect® will use a licensing business model for selective chemotherapeutic compounds to be developed by us.
Intellectual Property Portfolio
As of September 1, 2023, we and our subsidiary, OncoSelect®, have a patent estate that includes 15 issued U.S. and foreign counterpart patents, including two U.S. patents and thirteen foreign counterpart patents in Australia, Canada, China, France, Germany, Hong Kong, Italy, Mexico, Spain, Sweden, and the United Kingdom. One U.S. patent and nine counterpart foreign patents directed at diagnostic applications expire in 2030. Therapeutic patents registered in Australia, China, Hong Kong, Mexico, and the United States expire in 2037.
With regard to our diagnostic test CyPath® Lung and other diagnostic candidates, we have one issued U.S. patent and nine foreign counterpart patents in Canada, China, France, Germany, Hong Kong, Italy, Spain, Sweden, and the United Kingdom. With regard to our diagnostic patent applications, one of two families is directed at diagnosing lung health using flow cytometry, and the other is directed at proprietary compensation beads used to calibrate the flow cytometry instrument and used in CyPath® Lung data acquisition. Pending applications directed at diagnosing lung health include one pending U.S. non-provisional patent application and eight foreign counterpart patent applications in Australia, Canada, China, European Patent Office, Hong Kong, Japan, Mexico, and Singapore filed in 2019, one International Patent Application filed in 2022 and one International Patent Application filed in 2023. Also, a patent application directed at the composition of compensation beads was filed as an International Patent Application in 2022.
With regard to our therapeutic product candidates, we have one issued U.S. patent, four issued foreign patents, two pending U.S. applications, nine foreign applications pending in Canada, China, European Patent Office, Hong Kong, India, and Japan and one pending International Patent Application filed in 2022. The therapeutic intellectual property is made up of two families directed at our therapeutic product candidates, including one family directed at siRNA product candidates for the treatment of cancer, and another family directed at porphyrin conjugates for treating cancer.
| 6 |
Industry Opportunity
The global market for cancer diagnostic tests is expected to grow dramatically in coming years. According to a 2021 Global Cancer Diagnostics Market Research Report, cancer diagnostic tests, including devices, grew from $156.27 billion in 2020 to $170.21 billion in 2021, with a compound annual growth rate of 8.9%, and is projected to reach $239.23 billion in 2025. Lung cancer is the most common cancer globally and its incidence continues to increase in some large nations including China, where lung cancer is the leading cause of cancer-related morbidity and mortality, as reported in the Journal of Thoracic Oncology in October 2020 in an article entitled “Lung Cancer in People’s Republic of China.” According to a 2023 report “Lung Cancer Diagnostics: Global Strategic Business Report,” the global market for lung cancer diagnostic tests was estimated at $2.6 billion in 2022 and is projected to reach a value of $4.7 billion by 2030, with a compounded annual growth rate of 7.8% over 2022-2030. Clinical diagnostics play an important role in disease prevention, detection, and management. Our first test, CyPath® Lung, focuses on the leading cause of cancer death among both men and women. Lung cancer accounted for approximately 18% of all cancer deaths worldwide in 2020, as reported by the World Health Organization. Lung cancer typically may not be symptomatic in its early stages when it is most treatable. An estimated 14 million patients at high risk for lung cancer in the U.S. are recommended for annual screening. Initially, physicians would order CyPath® Lung for those high-risk patients as an adjunct to LDCT screening to aid in the decision whether to pursue more aggressive follow-up procedures. A more accurate and reliable lung diagnostic pathway using LDCT and noninvasive methods could result in fewer patients being subjected to the stresses of unnecessary, invasive diagnostic procedures such as biopsies. CyPath® Lung is well suited for use in both sophisticated and less-developed markets because sample collection is noninvasive and conducted at home, the sample can be shipped overnight by commercial carriers and sample processing and automated analysis can be completed by laboratory technicians skilled in general laboratory techniques. Patient reports are provided to the ordering physician within three days of sample receipt at the laboratory.
Competitive Strengths
We conduct an ongoing competitive analysis of companies in the lung cancer diagnostic sector of the clinical diagnostics market. In 2022, we evaluated companies that reported an interest in diagnosing lung cancer, focusing on 67 companies and academic institutions we identified as active in the early lung cancer diagnostic sector. A thorough evaluation of the early lung cancer diagnostic landscape reveals multiple reasons why CyPath® Lung is positioned to be a market leader. CyPath® Lung performance shown in our test validation trial resulted in 92% sensitivity and 87% specificity in high-risk patients who had lung nodules 20 mm or smaller. Eight out of 10 (80%) Stage I tumors were correctly identified, indicating that CyPath® Lung can find lung cancer at its earliest stage. Overall, when diagnosing lung cancer in all stages, the clinical trial resulted in CyPath® Lung specificity of 88% and sensitivity of 82%, similar to far more invasive procedures and surgery currently used to diagnose lung cancer. (See the “Comparison of CyPath® Lung to Current Standards of Care” chart in the “Business” section of this prospectus.) The test validation trial of 150 patients was conducted over 19 months. Participants provided a sputum sample and were released from the study after a physician either confirmed the individual was cancer-free by examination of CT imaging or confirmed the presence of lung cancer by biopsy. Test data used to produce results included: (1) the proportion of cells with a high ratio of high TCPP fluorescence intensity over cell size; (2) the proportion of cells with an intermediate ratio of fluorescence intensity caused by the viability dye (FVS510) over cell size; (3) the proportion of cells that were CD206 negative but positive for one or more of the following markers: CD66b (granulocytes), CD3 (T cells), and CD19 (B cells); and (4) patient age.
The majority of competitors’ tests either incorrectly classify a high proportion of people without cancer as having the disease (known as false negatives) more than 50% of the time or misdiagnose people as cancer-free (known as false positives) more than 50% of the time. It is important to note that most competitors who have conducted clinical trials also have not designed their trials to evaluate the test’s measure of accuracy – such as sensitivity and specificity – in the high-risk population for whom the test is intended A patient collects his or her sample at home, which is a particular benefit during a pandemic. Sample processing for CyPath® Lung can be done by laboratory technicians, and reagents used by the test are widely available. Data acquisition and analysis and test results are fully automated.
Business Strategies
We are moving forward with commercialization of CyPath® Lung in a systematic, four-phased business plan (“Business Plan”) for market entry into the U.S., the European Union (“EU”), and worldwide that are timed to maximize Company resources and minimize market risk. Phase 1 of our Business Plan has begun with a limited market launch of the Company’s CyPath® Lung LDT in South Texas. This limited test market launch is designed to evaluate our marketing program and help us ensure each step in the care pathway – from the initial order by physicians to sputum collection and processing, to generating and delivering the patient report – is efficient and effective. This limited test market approach allows us to refine future positioning and develop strategic insight for our CyPath® Lung test before expanding to a larger market. The next step in our marketing plan provides for and will be followed by expansion into the Southwest market area in 2024 followed by a staged nationwide expansion of sales and marketing. Phase 2 of our Business Plan anticipates entering the EU market with CyPath® Lung as a CE-marked in vitro diagnostic (“IVD”) test with sales in the Netherlands, followed by a staged EU expansion. Phase 3 of our Business Plan focuses on the marketing of an FDA-cleared CyPath® Lung test, beginning with a pivotal clinical trial in the U.S.
In Phase 3, we plan to submit a request for de novo classification to the FDA to classify CyPath® Lung as a Class II IVD medical device for the detection of lung cancer. In order to seek de novo classification and marketing authorization of CyPath® Lung by the FDA, we must conduct a “pivotal clinical trial” to demonstrate the safety and efficacy of CyPath® Lung. We are currently working with a contract research organization (“CRO”) to finalize the protocol for the pivotal clinical trial and plan to submit a pre-submission package to the FDA in the fourth quarter of 2023 to obtain the FDA’s feedback on the study design. A pivotal clinical trial is scheduled to begin in early 2024. Final design of the pivotal clinical trial has not been determined at this time; however we estimate enrollment of approximately 1,800 participants at high risk for lung cancer and expect the trial to require three years. Assuming the study is successful, we intend to submit a de novo classification request to the FDA within six months of study completion. If the de novo request is granted by the FDA, we expect such marketing authorization to result in a larger market and greater market share for CyPath® Lung. FDA marketing authorization also can lead to expanded claims and additional indications for use of CyPath® Lung for the early detection of lung cancer. Phase 4 will accelerate the diagnostic’s market presence to expand into other global markets, including China, Southeast Asia, and Australia.
Corporate Information
We were incorporated in the State of Delaware on March 26, 2014. Our principal executive office is located at 22211 West Interstate 10, Suite 1206, San Antonio, Texas 78257, and our telephone number at that address is (210) 698-5334. Our website address is https://www.bioaffinitytech.com/. Information contained on or that can be accessed through our website is not incorporated by reference into this prospectus. Investors should not consider any such information to be part of this prospectus.
| 7 |
Implications of Being an Emerging Growth Company
We qualify as an “emerging growth company” (an “EGC”) as defined in the Jumpstart Our Business Startups Act of 2012 (the “JOBS Act”). As an EGC, for up to five years, we may elect to take advantage of certain specified exemptions from reporting and other regulatory requirements that are otherwise generally applicable to public companies. For example, these exemptions would allow us to:
| ● | present two, rather than three, years of audited financial statements with correspondingly reduced disclosure in the “Management’s Discussion and Analysis of Financial Condition and Results of Operations” section (the “MD&A”) of this prospectus; | |
| ● | defer the auditor attestation requirement on the effectiveness of our system of internal control over financial reporting; | |
| ● | make reduced disclosures about our executive compensation arrangements; and | |
| ● | forego the adoption of new or revised financial accounting standards until they would be applicable to private companies. |
Certain of these reduced reporting requirements and exemptions were already available to us due to the fact that we also qualify as a “smaller reporting company” under SEC rules. For instance, smaller reporting companies are not required to obtain an auditor attestation and report regarding internal control over financial reporting, to provide a compensation discussion and analysis, or to provide a pay-for-performance graph or CEO pay ratio disclosure, and they may present two, rather than three, years of audited financial statements and related MD&A disclosure.
We may take advantage of these exemptions up until the last day of the fiscal year following the fifth anniversary of our initial public offering or until we are no longer an EGC, which would be the case if (i) our total annual gross revenues are $1.235 billion or more; (ii) we issue more than $1 billion in non-convertible debt during a consecutive three-year period; or (iii) we become a “large accelerated filer,” as defined in the Securities Exchange Act of 1934, as amended (the “Exchange Act”). We may choose to take advantage of some, but not all, of the available exemptions. We have taken advantage of certain reduced reporting obligations in this prospectus. Accordingly, the information contained herein may be different than the information you receive from other public companies in which you hold stock. For more information, see “Risk Factors—General Risk Factors—We are an “emerging growth company,” and the reduced disclosure requirements applicable to emerging growth companies may make our Common Stock less attractive to investors.”
Summary of Risk Factors
Like any emerging growth company, we face significant risk factors that may impede our plans for successful commercialization of our diagnostic and therapeutic products. These risks are discussed in detail under the “Risk Factors” discussion beginning on page 14 of this prospectus.
The following summarizes the principal factors that make an investment in our Company speculative or risky, all of which are more fully described in the section below titled “Risk Factors.” This summary should be read in conjunction with the section below titled “Risk Factors” and should not be relied upon as an exhaustive summary of the material risks facing our business. The following factors could result in harm to our business, reputation, revenue, financial results, and prospects, among other impacts:
| ● | We may not experience the anticipated strategic benefits of the Acquisition; | |
| ● | We may be unable to successfully integrate the clinical pathology laboratory business with ours; | |
| ● | The future revenue to be generated from PPLS is uncertain; | |
| ● | The market price of our common stock following the Acquisition may decline; | |
| ● | Our stockholders will experience substantial dilution from the issuance of the Acquisition consideration; |
| ● | our limited operating history and history of net losses since our inception; | |
| ● | our need to obtain substantial additional funding to complete the development and commercialization of our diagnostic tests and therapeutic product candidates; | |
| ● | potential dilution to our stockholders, including purchasers of Common Stock in this Offering, resulting from the conversion of our preferred stock, par value $0.001 per share (our “Preferred Stock”) and convertible debt outstanding, and potential restrictions, due to raising additional capital; | |
| ● | the impact of a material weakness identified in our internal control over financial reporting; | |
| ● | the early stage of our development efforts; | |
| ● | the unpredictability of future trial results; | |
| ● | the difficulty in predicting the results, timing, and cost of our development of our diagnostic tests and therapeutic product candidates and the likelihood of obtaining regulatory approval; | |
| ● | the risk of experiencing delays or difficulties in the enrollment and/or retention of patients in clinical trials; |
| 8 |
| ● | potential changes to interim, “top-line” or preliminary results from our clinical trials as more patient data becomes available and are subject to audit and verification procedures; | |
| ● | the risk that the FDA may not agree with our LDT regulatory strategy or that Congress may enact legislation giving the FDA new authorities to regulate LDTs; | |
| ● | the lengthy, time-consuming, and unpredictable nature of regulatory approval processes; | |
| ● | the risk that our preclinical studies and clinical trials fail to demonstrate the safety and efficacy of our diagnostic tests or therapeutic product candidates; | |
| ● | the risk that data from clinical trials conducted outside of the United States may not be accepted by regulatory authorities; | |
| ● | the impact of ongoing regulatory obligations and continued regulatory review, even if we receive regulatory approval for any of our diagnostic tests or therapeutic product candidates; | |
| ● | our lack of control over the supply, regulatory status, or regulatory approval of third-party drugs or biologics with which our diagnostic tests or therapeutic product candidates are used in combination; | |
| ● | our lack of control over the conduct of investigator-initiated clinical trials or other clinical trials sponsored by organizations or agencies other than us; | |
| ● | the risk that we fail to develop additional diagnostic tests or therapeutic product candidates; | |
| ● | the risk that we are unable to penetrate multiple markets; | |
| ● | the risk that our diagnostic tests and therapeutic product candidates may fail to achieve market acceptance, even if they receive marketing authorization; | |
| ● | if we are unable to obtain and maintain sufficient intellectual property protection for our platform and our diagnostic tests or therapeutic product candidates, or if the scope of the intellectual property protection is not sufficiently broad, our competitive position may be adversely affected; | |
| ● | the price of our stock may be volatile, and you could lose all or part of your investment. Unstable market and economic conditions may have serious adverse consequences on our business, financial condition and stock price; | |
| ● | our success is highly dependent on our ability to attract and retain highly skilled executive officers and employees; | |
| ● | we face significant competition from other biotechnology and pharmaceutical companies, and our operating results will suffer if we fail to compete effectively; and | |
| ● | our business is affected by the ongoing COVID-19 pandemic and may be significantly adversely affected as the pandemic continues or if other events out of our control disrupt our business or that of our third-party providers. |
| 9 |
THE OFFERING
| Issuer | bioAffinity Technologies, Inc. | |
| Securities Offered | 3,086,419 Units (based on an assumed public offering price of $1.62 per Unit, which is based on the closing price of the Common Stock on September 13, 2023), each Unit consisting of one share of our Common Stock and one Warrant. | |
| Description of the Warrants | Each Warrant is exercisable for one share of Common Stock for an assumed price of $[●] per share (120% of the Offering Price of one Unit). Each Warrant will be exercisable immediately upon issuance and will expire five (5) years after the initial issuance date. For more information regarding the Warrants, you should carefully read the section titled “Description of Securities—Warrants” on page 99 of this prospectus. | |
| Over-Allotment Option | We have granted the underwriters a 45-day Over-Allotment Option to purchase up to an additional 462,962 shares of Common Stock and /or 462,962 additional Warrants at a per share price equal to the Offering Price per Unit minus $0.01 and per Warrant price of $0.01, or any combination of additional shares of Common Stock and Warrants, in all cases less the underwriting discounts payable by us. | |
Shares of Common Stock Outstanding prior to the Offering(1) |
9,350,297 shares as of September 20, 2023 |
|
| Shares of Common Stock Outstanding after the Offering(1)(2) | 12,436,716 shares | |
| Use of Proceeds |
We estimate that the net proceeds to us from the sale of our Units in this Offering will be approximately $4.0 million, after deducting the underwriting discounts and commissions and estimated offering expenses payable by us. If the underwriters exercise their Over-Allotment Option in shares of Common Stock in full, the net proceeds to us will be approximately $4.7 million.
We intend to use the net proceeds from this Offering for working capital and for general corporate purposes, which may include laboratory test and therapeutic product development, general and administrative matters, and capital expenditures. We may also use a portion of the net proceeds for the acquisition of, or investment in, technologies, solutions or businesses that complement our business, although we have no present definitive commitments or agreements to enter into any acquisitions or investments.
We cannot specify with certainty all of the uses of the net proceeds that we will receive from this Offering. Accordingly, we will have broad discretion in the application of these proceeds and our investors will be relying on the judgment of our management regarding the application of the net proceeds of this Offering. |
|
| Representative’s Warrant | The registration statement of which this prospectus is a part also registers for sale the Representative’s Warrant, which gives the Representative the right to purchase up to 2.0% (subject to reduction) of the shares of Common Stock underlying the Units sold in this Offering, as a portion of the underwriting compensation in connection with this Offering. The Representative’s Warrant will be exercisable at any time, and from time to time, in whole or in part, commencing on a date that is 180 days after the commencement of sales of the Units in this Offering and expiring five years from the date of the registration statement in this Offering at an exercise price of $[●] (120% of the assumed offering price per Unit). We are registering the Representative’s Warrant and the shares of Common Stock underlying the Representative’s Warrant in the registration statement of which this prospectus is a part. See “Underwriting—Representative’s Warrant” on page 108 of this prospectus for a description of the Representative’s Warrant. |
| 10 |
| Lock-Up Agreements | We have agreed with the underwriters not to sell additional equity securities for a period of 180 days after the effective date of this Offering. Our directors and officers have agreed with the underwriters not to offer for sale, issue, sell, contract to sell, pledge or otherwise dispose of any of our Common Stock or securities convertible into Common Stock, subject to certain exceptions, for a period of 180 days after the date of this prospectus, which restriction may be waived in the discretion of the Representative. See “Underwriting—Lock-Up Agreements” on page 108 of this prospectus. | |
| Risk Factors | You should read the “Risk Factors” section beginning on page 14 of this prospectus and the other information included herein for a discussion of factors to consider prior to deciding to invest in our Units. | |
| Nasdaq Capital Market Listing | Our Common Stock is listed on the Nasdaq Capital Market under the symbol “BIAF and our Warrants issued in our initial public offering are listed on the Nasdaq Capital Market under the symbol “BIAFW.” | |
| Transfer Agent and Warrant Agent | The transfer agent and registrar for our Common Stock and the Warrant Agent for our Warrants is VStock Transfer, LLC. |
| (1) | The number of shares of Common Stock outstanding immediately prior to and after this Offering is based on 9,350,297 shares of Common Stock outstanding as of September 20, 2023 and includes: (i) 133,414 unvested shares of restricted Common Stock, which are subject to forfeiture, but which are outstanding and have voting rights; and (ii) 564,972 shares of Common Stock issued to the Joyce Trust pursuant to the terms of the Asset Purchase Agreement on September 20, 2023. |
| (2) |
The number of shares of Common Stock outstanding immediately following this Offering is based on all of the shares listed in number (1) above and excludes: |
| ● | 3,086,419 shares of Common Stock issuable upon the exercise of the Warrants underlying the Units sold in this Offering; | |
| ● | 462,962 shares of Common Stock issuable upon the exercise of the Over-Allotment Option; | |
| ● | 462,962 shares of Common Stock issuable upon the exercise of 462,962 Warrants issuable upon the exercise of the Over-Allotment Option; | |
| ● | 61,728 shares of Common Stock issuable upon the exercise of the Representative’s Warrant; | |
| ● | 806,392 shares of Common Stock issuable upon the exercise of stock options issued under our 2014 Equity Incentive Plan to certain of our employees, directors, and consultants with a weighted average exercise price equal to $4.33; | |
| ● | an aggregate of 4,305,812 shares of Common Stock issuable upon the exercise of outstanding Tradeable Warrants and Non-Tradeable Warrants, (which share number reflects an adjustment in the number of shares to be issued upon exercise of the Tradeable Warrants and Non-Tradeable Warrants that will be effected upon consummation of this Offering in accordance with the price protection provisions contained in the Tradeable Warrants and Non-Tradeable Warrants) all of which, upon consummation of the Acquisition, will have an exercise price that will be reduced to $3.0625 per share; | |
| ● | 2,900,904 shares of Common Stock issuable upon the exercise of outstanding warrants issued prior to consummation of our initial public offering, with a weighted average exercise price equal to $5.31 per share; and | |
| ● | 658,294 shares of our Common Stock that are reserved for equity awards that may be granted under our 2014 Equity Incentive Plan. |
Except as otherwise indicated, all information in this prospectus assumes:
| ● | An assumed initial Offering Price of $1.62 per Unit, which is the closing price of our Common Stock on the Nasdaq on September 13, 2023 set forth on the cover page of this prospectus; | |
| ● | no exercise of the Warrants underlying the Units in this Offering; | |
| ● | no exercise of any options under our 2014 Equity Incentive Plan; | |
| ● | no exercise of any outstanding warrants; and | |
| ● | no exercise of the Over-Allotment Option. |
| 11 |
SUMMARY FINANCIAL DATA
We have derived the following summary of our consolidated statement of operations data for the years ended December 31, 2022 and 2021, and the consolidated balance sheet data as of December 31, 2022 and 2021, from our audited consolidated financial statements appearing elsewhere in this prospectus. We have derived the following summary of our condensed consolidated statement of operations data for the six months ended June 30, 2023 and 2022, and the balance sheet data as of June 30, 2023, from our unaudited interim condensed consolidated financial statements appearing elsewhere in this prospectus. The unaudited interim condensed consolidated financial statements have been prepared on a basis consistent with our audited consolidated financial statements included in this prospectus and include, in our opinion, all adjustments, consisting only of normal recurring adjustments, necessary for the fair statement of the financial information in those statements. Our historical results are not necessarily indicative of the results that may be expected in the future.
You should read the following summary financial data together with our consolidated financial statements and the related notes appearing elsewhere in this prospectus and the MD&A section of this prospectus.
The following table summarizes our results of operations for the years ended December 31, 2022 and 2021 and the six months ended June 30, 2023 and 2022.
| Year Ended | Six Months Ended | |||||||||||||||
| December 31, | June 30, | |||||||||||||||
| 2022 | 2021 | 2023 | 2022 |
|||||||||||||
| (Unaudited) | ||||||||||||||||
| Revenues | $ | 4,803 | $ | — | $ | 20,659 | 1,306 | |||||||||
| Cost of sales | 467 | — | 1,322 | 146 | ||||||||||||
| Gross profit | 4,336 | — | 19,337 | 1,160 | ||||||||||||
| Operating expenses | ||||||||||||||||
| Research and development | 1,142,777 | 1,007,476 | 704,741 | 528,267 | ||||||||||||
| Clinical development | 145,546 | 130,475 | 54,888 | 80,744 | ||||||||||||
| Selling, general and administrative | 2,727,071 | 1,068,871 | 2,596,027 | 803,311 | ||||||||||||
| Total operating expense | 4,015,394 | 2,206,822 | 3,355,657 | 1,412,322 | ||||||||||||
| Loss from operations | (4,011,058 | ) | (2,206,822 | ) | (3,336,320 | ) | (1,411,162 | ) | ||||||||
| Other income (expense), including tax | (4,143,055 | ) | (4,119,591 | ) | 96,169 | (144,592 | ) | |||||||||
| Net (loss) income | $ | (8,154,113 | ) | $ | (6,326,413 | ) | $ | (3,272,963 | ) | $ | (1,560,072 | ) | ||||
| Net (loss) income per common share: | ||||||||||||||||
| Basic and Diluted | $ | (1.81 | ) | $ | (2.36 | ) | $ | (0.38 | ) | $ | (0.58 | ) | ||||
| $ | $ | $ | ||||||||||||||
| Weighted average common shares outstanding: | ||||||||||||||||
| Basic and Diluted | 4,498,964 | 2,675,270 | 8,477,656 | 2,687,431 | ||||||||||||
The following table summarizes our consolidated balance sheets at June 30, 2023 (amounts in thousands):
| As of | ||||||||||||
| June 30, 2023 | ||||||||||||
| (Unaudited) | ||||||||||||
| Actual(1) | Pro Forma(1) | Pro Forma, As Adjusted(2) | ||||||||||
| Cash and cash equivalents | $ | 8,279,182 | 6,161,830 | $ | 10,151,830 | |||||||
| Working capital (deficit) | $ | 7,884,052 | 6,298,838 | $ | 10,288,838 | |||||||
| Total assets | $ | 8,873,499 |
12,064,004 |
$ | 16,054,004 | |||||||
| Total liabilities | $ | 775,151 |
2,965,656 |
$ | 2,965,656 |
|||||||
| Accumulated deficit | $ | (39,940,431 | ) | (40,111,473 | ) | $ | (40,111,473 | ) | ||||
| Total stockholders’ equity (deficit) | $ | 8,098,348 |
9,098,348 |
$ | 13,088,348 |
|||||||
| (1) | The actual and pro forma balance sheet data in the table above does not include unvested shares of restricted Common Stock (which was 133,414 as of September 20, 2023, and the pro forma balance sheet data gives effect to: (i) on July 1, 2023, the issuance of an aggregate of 71,715 restricted shares of Common Stock to our seven directors, as part of our director compensation policy; (ii) the issuance of an aggregate of 8,226 shares of Common Stock to a consultant pursuant to the terms of a consulting agreement between July 1, 2023 and September 1, 2023; (iii) the addition of 16,605 shares of restricted Common Stock that were issued but unvested prior to June 30, 2023 and which have subsequently vested; and (iv) the consummation of the Acquisition, including the issuance of 564,972 shares of restricted Common Stock to the Joyce Trust pursuant to the terms of the Asset Purchase Agreement, the payment of $2,500,000 cash consideration to Village Oaks, the assets acquired including cash and the liabilities assumed in Acquisition. |
| (2) | The pro forma, as adjusted balance sheet data in the table above reflects the items described in footnote (1) above and gives effect to the issuance and sale of Units in this Offering at an assumed Offering Price of $1.62 per Unit, after deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us. |
| 12 |
Cautionary Note Regarding Forward-Looking Statements
| ● | our projected financial position and estimated cash burn rate; | |
| ● | our estimates regarding expenses, future revenues and capital requirements; | |
| ● | our ability to obtain funding for our operations necessary to complete further development and commercialization of our diagnostic tests or therapeutic product candidates; | |
| ● | our dependence on third parties in the conduct of our clinical trials; | |
| ● | our ability to obtain the necessary regulatory approvals to market and commercialize our diagnostic tests or therapeutic product candidates; | |
| ● | the potential that the results of our pre-clinical and clinical trials indicate our current diagnostic tests or any future diagnostic tests or therapeutic product candidates we may seek to develop are unsafe or ineffective; | |
| ● | the results of market research conducted by us or others; | |
| ● | our ability to obtain and maintain intellectual property protection for our current diagnostic tests or future diagnostic and therapeutic product candidates; | |
| ● | our ability to protect our IP rights and the potential for us to incur substantial costs from lawsuits to enforce or protect our IP rights; | |
| ● | the possibility that a third party may claim we or our third-party licensors have infringed, misappropriated, or otherwise violated their IP rights and that we may incur substantial costs and be required to devote substantial time defending against such claims; | |
| ● | our reliance on third parties; | |
| ● | the success of competing therapies, diagnostic tests, and therapeutic products that are or will become available; | |
| ● |
our ability to expand our organization to accommodate potential growth and to retain and attract key personnel;
|
|
| ● | our potential to incur substantial costs resulting from product liability lawsuits against us and the potential for such lawsuits to cause us to limit the commercialization of our diagnostic tests and therapeutic product candidates; | |
| ● | market acceptance of our diagnostic tests and therapeutic product candidates, the size and growth of the potential markets for our current diagnostic tests and therapeutic product candidates, and any future diagnostic tests and therapeutic product candidates we may seek to develop, and our ability to serve those markets; | |
| ● | the successful development of our commercialization capabilities, including sales and marketing capabilities; | |
| ● | compliance with government regulations, including environmental, health, and safety regulations and liabilities thereunder; | |
| ● |
the impact of any health epidemic, on our business, our clinical trials, our research programs, healthcare systems, or the global economy as a whole;
|
|
| ● | general instability of economic and political conditions in the United States, including inflationary pressures, increased interest rates, economic slowdown or recession, and escalating geopolitical tensions; |
|
| ● | compliance with government regulations, including environmental, health, and safety regulations and liabilities thereunder; | |
| ● | our anticipated uses of net proceeds from this offering; | |
| ● | the increased expenses associated with being a public company; and | |
| ● | other factors discussed elsewhere in this prospectus. |
Many of the foregoing risks and uncertainties, as well as risks and uncertainties that are currently unknown to us, are or may be exacerbated by factors such as the ongoing conflict between Ukraine and Russia, escalating tensions between China and Taiwan, increasing economic uncertainty and inflationary pressures, and the emergence of new viral variants, and any consequent worsening of the global business and economic environment. New factors emerge from time to time, and it is not possible for us to predict all such factors. Should one or more of the risks or uncertainties described in this Annual Report or any other filing with the Securities and Exchange Commission (the “SEC”) occur, or should the assumptions underlying the forward-looking statements we make herein and therein prove incorrect, our actual results and plans could differ materially from those expressed in any forward-looking statements. We undertake no obligation to update publicly any forward-looking statements, whether as a result of new information, future events, or otherwise, except as required by law.
In addition, statements such as “we believe” and similar statements reflect our beliefs and opinions on the relevant subject. These statements are based upon information available to us as of the date of this prospectus and, although we believe such information forms a reasonable basis for such statements, such information may be limited or incomplete, and our statements should not be read to indicate that we have conducted a thorough inquiry into, or review of, all potentially available relevant information. These statements are inherently uncertain and investors are cautioned not to unduly rely upon these statements. Furthermore, if our forward-looking statements prove to be inaccurate, the inaccuracy may be material. In light of the significant uncertainties in these forward-looking statements, you should not regard these statements as a representation or warranty by us or any other person that we will achieve our objectives and plans in any specified time frame, or at all. Except as required by applicable law, we do not plan to publicly update or revise any forward-looking statements contained herein until after we distribute this prospectus, whether as a result of any new information, future events or otherwise.
You should not place undue reliance on our forward-looking statements. Although forward-looking statements reflect our good-faith beliefs at the time they are made, forward-looking statements involve known and unknown risks, uncertainties, and other factors, including the factors described under “Risk Factors,” which may cause our actual results, performance or achievements to differ materially from anticipated future results, performance, or achievements expressed or implied by such forward-looking statements. We undertake no obligation to publicly update or revise any forward-looking statement, whether as a result of new information, future events, changed circumstances, or otherwise, unless required by law. These cautionary statements qualify all forward-looking statements attributable to us or persons acting on our behalf.
| 13 |
RISK FACTORS
Investing in our Company involves a high degree of risk. You should carefully consider the following information about these risks, together with the other information appearing elsewhere in this Prospectus before deciding to invest in our Company. The occurrence of any of the following risks could have a material and adverse effect on our business, reputation, financial condition, results of operations, and future growth prospects, as well as our ability to accomplish our strategic objectives. As a result, the market value of our Common Stock could decline, and you could lose all or part of your investment. Additional risks and uncertainties not presently known to us or that we currently deem immaterial may also impair our business operations and market value.
Risks Related to the Acquisition
The combined company may not experience the anticipated strategic benefits of the Acquisition.
While we anticipate benefits from the Acquisition, we may not be able to realize the expected benefits. We may not be able to integrate the two businesses successfully, and despite due diligence we could assume previously unidentified or contingent liabilities. Ownership of a CAP/CLIA laboratory and related services business may not have the clinical value and commercial potential which we envision. Any substantive failure of the Acquisition to meet our expectations could have a material negative effect on our results of operations. There can be no assurance that the anticipated benefits of the Acquisition will materialize or that if they materialize will result in increased stockholder value or revenue stream to the combined company.
We may be unable to successfully integrate the PPLS business with our current management and structure.
Our failure to successfully complete the integration of PPLS could have an adverse effect on our prospects, business activities, cash flow, financial condition, results of operations and stock price. Integration challenges may include the following:
| ● | assimilating and retaining former Village Oaks personnel who joined PPLS as part of the Acquisition; | |
| ● | estimating the capital, personnel and equipment required for the operation of PPLS based on the historical experience of management with the businesses they are familiar with; and | |
| ● | minimizing potential adverse effects on existing business relationships. |
We may not be able to enforce claims with respect to the representations, warranties and indemnities that Village Oaks has provided to us under the Asset Purchase Agreement.
In connection with the Acquisition, Village Oaks has given certain representations, warranties and indemnities. There can be no assurance we will be able to enforce any claims against Village Oaks’ breaches of such representations, warranties or indemnities. Village Oaks’ liability with respect to breaches of such representations, warranties and indemnities under the Asset Purchase Agreement may be limited or the amount and coverage of any insurance obtained with respect to representations and warranties may be limited. Even if we ultimately succeed in recovering any amounts, we may temporarily be required to bear these losses ourselves.
We are unable to precisely estimate when we will begin to generate significant profit from revenue, if ever, from PPLS’ services, nor to estimate the amount of profit or revenue that will be generated or the expenses that will be incurred.
We do not expect to immediately derive profit from revenue from PPLS’ services. Once we begin to generate such profit, there is no guarantee that it will be sufficient to realize the expected financial benefits of the Acquisition. In addition, since we have limited experience operating a clinical laboratory, we may not accurately estimate the expenses we will incur.
| 14 |
The market price of our common stock following the Acquisition may decline as a result of such Acquisition.
The market price of our common stock may decline as a result of the Acquisition for a number of reasons including if:
| ● | investors react negatively to the prospects of our business after the Acquisition; | |
| ● | the effect that the Acquisition has on our business and prospectus is not consistent with the expectations of financial or industry analysists; or | |
| ● | after the Acquisition, the Company does not achieve the perceived benefits of the Acquisition as rapidly or to the extent anticipated by financial or industry analysts. |
Operating a clinical laboratory is a new business for us and the members of our management team have limited experience operating a CAP-accredited, CLIA-certified laboratory, which may limit the ability of investors to make an informed investment decision.
We have never operated a clinical laboratory. To date, only our Chief Operating Officer, Xavier Reveles, has operated a CAP-accredited, CLIA-certified clinical laboratory and therefore it may be difficult for investors to analyze our ability to successfully operate a clinical laboratory. Our management team may not successfully or efficiently manage our transition to operating a CAP-accredited and CLIA-certified laboratory subject to significant regulatory oversight and reporting obligations. However, to ease the transition, Dr. Joyce, the Medical Director and Laboratory Director of Village Oaks prior to the Acquisition, continues to serve as the Medical Director and Laboratory Director of PPLS. These new obligations and constituents will require significant attention from our senior management and could divert their attention away from the day-to-day management of our business, which could adversely affect our business, financial condition, and operating results.
Our stockholders will experience substantial dilution from the issuance of the consideration paid in connection with the Acquisition and may not realize a benefit from the Acquisition commensurate with the ownership dilution they will experience in connection with the Acquisition.
Our stockholders will experience substantial dilution from the issuance of the consideration paid in connection with the Acquisition. If after the Acquisition we are unable to realize the full strategic and financial benefits currently anticipated from the Acquisition, our securityholders will have experienced substantial dilution of their ownership interests without receiving any commensurate benefit, or only receiving part of the commensurate benefit to the extent the post-acquisition company is able to realize only part of the strategic and financial benefits currently anticipated from the Acquisition.
The unaudited pro forma financial information included in this prospectus is for illustrative purposes and the combined company’s actual financial position or results of operations after the anticipated Acquisition may differ materially.
The unaudited pro forma financial information in this prospectus is presented for illustrative purposes only and is not necessarily indicative of what the combined company’s actual financial position or results of operations would have been had the Acquisition been completed on the dates indicated. The unaudited pro forma financial information reflects adjustments, which are based upon estimates, to allocate the purchase price to tangible and identifiable intangible assets acquired and liabilities assumed, based on their estimated acquisition-date fair values. The purchase price allocation reflected in this document is preliminary, and a final determination of the fair value of assets acquired and liabilities assumed will be based on the actual net tangible and intangible assets and liabilities of Village Oaks that existed as of the date on which the Acquisition was consummated. Accordingly, the final purchase accounting adjustments may differ materially from the pro forma information reflected in this prospectus. For more information, please see the section entitled “bioAffinity Technologies, Inc. Unaudited Pro Forma Combined Financial Statements.”
| 15 |
Risks Related to Our Financial Position
Our Business Plan relies upon our ability to obtain additional sources of capital and financing. If the amount of capital we are able to raise from financing activities, together with our revenues from operations, is not sufficient to satisfy our capital needs, we may be required to cease operations.
Prior to 2022, we had not generated any revenue. During the year ended December 31, 2022, we generated approximately $5,000 and during the six months ended June 30, 2023 we generated approximately $8,000 in revenue from royalties from sales of our first diagnostic test, CyPath® Lung by Village Oaks, a CAP-accredited, CLIA-certified clinical pathology laboratory to which we had previously granted a license to develop CyPath® Lung for commercialization and to manufacture, use, market and sell CyPath® Lung as an LDT prior to the Acquisition, which license was assigned to and assumed by PPLS in connection with the Acquisition, that began a limited market launch in the second quarter of 2022 to pulmonologists in South Texas. During the six months ended June 30, 2023, we also generated revenue from clinical flow cytometry services provided to Village Oaks related to CyPath® Lung in the approximate amount of $3,000 and in connection with CyPath® Lung tests purchased by the U.S. Department of Defense in the approximate amount of $10,000 for an observational study.
To become and remain profitable, we must succeed in developing and commercializing our diagnostic tests and therapeutic products that we expect will generate significant income in the planned timeframe. This will require us to be successful in a range of challenging activities, including completing preclinical testing and clinical trials of our diagnostic and therapeutic technologies, obtaining regulatory approval for our diagnostic and therapeutic technologies, manufacturing, marketing and selling any diagnostic tests and therapeutic products for which we may obtain regulatory approval, and establishing and managing our collaborations at various phases of each diagnostic test and therapeutic product candidate’s development. We are in the preliminary phases of these activities. We may never succeed in these activities and, even if we do, may never generate sufficient income to achieve profitability.
To become profitable, we must develop our diagnostic tests and therapeutic products, which will depend in large part on our ability to:
| ● | Develop, enhance and protect our diagnostic tests and therapeutic products; | |
| ● | Raise sufficient funding to support our diagnostic tests and therapeutic product development program(s); | |
| ● | Complete pre-clinical testing; | |
| ● |
Work with our partners to expand commercialization of our first diagnostic test, CyPath® Lung, as an LDT under the CAP/CLIA guidelines and regulations administered by CMS and CAP; |
|
| ● | Obtain de novo classification from FDA for our CyPath® Lung as a Class II in vitro diagnostic | |
| ● | Work with our partners to develop and commercialize our first diagnostic test, CyPath® Lung, as a CE -marked test in accordance with the In Vitro Diagnostic Device Regulation (the “IVDR”) of the EU; | |
| ● | Synthesize, test, and attract licensing partners for drug conjugates, siRNAs, and other therapeutics (and methods for their use) developed by us; | |
| ● | Develop and conduct human clinical studies to support the regulatory approval and marketing of our diagnostic test(s) and therapeutic product(s); | |
| ● | Develop and manufacture the test(s) and product(s) to FDA standards, appropriate EU standards, and appropriate standards required for the commercialization of our tests and products in countries in which we seek to sell our diagnostic test(s) and therapeutic product(s); | |
| ● | Obtain the necessary regulatory approvals to market our diagnostic test(s) and therapeutic product(s); | |
| ● | Secure the necessary personnel and infrastructure to support the development, commercialization, and marketing of our diagnostic test(s) and therapeutic product(s); and | |
| ● | Develop strategic relationships to support development, manufacturing, and marketing of our diagnostic test(s) and therapeutic product(s). |
Even if we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis. Our failure to become and remain profitable would depress the value of our Company and could impair our ability to raise capital, expand our business, maintain the research and development efforts that will be initially funded by the proceeds of this Offering, diversify our diagnostic tests and therapeutic product offerings, or even continue our operations. A decline in the value of our Company could also cause you to lose all or part of your investment.
We must raise additional capital to fund our operations in order to continue as a going concern.
As of December 31, 2022, we had an accumulated deficit of $36.7 million. As of June 30, 2023, we had an accumulated deficit of $39.9 million. Although we believe we will have sufficient cash on hand to fund our ongoing operations for a period of a least 12 months subsequent to the issuance of the unaudited condensed consolidated financial statements for the six months ended June 30, 2023, we need to raise further capital through the sale of additional equity or debt securities or other debt instruments, strategic relationships or grants, or other arrangements to support our future operations. Our Business Plan includes expansion for our commercialization efforts which will require additional funding. If we are unable to improve our liquidity position we may not be able to continue as a going concern. Our ability to continue as a going concern is dependent upon our ability to generate revenue and raise capital from financing transactions. Without funding from the proceeds of this Offering, management anticipates that our cash resources are sufficient to continue operations through May 2024. Our future is dependent upon its ability to obtain financing and upon future profitable operations from the development of its new business opportunities. There can be no assurance that we will be successful in accomplishing these objectives. Without such additional capital, we may be required to curtail or cease operations and be required to realize our assets and discharge our liabilities other than in the normal course of business which could cause investors to suffer the loss of all or a substantial portion of their investment. WithumSmith+Brown, PC, our independent registered public accounting firm for the fiscal year ended December 31, 2022, has included an explanatory paragraph in its opinion that accompanies our audited consolidated financial statements as of and for the year ended December 31, 2022, indicating that our current liquidity position raises substantial doubt about our ability to continue as a going concern.
| 16 |
We have a limited operating history, which makes it difficult to evaluate our current business and future prospects.
We are a company with limited operating history, and our operations are subject to all of the risks inherent in establishing a new business enterprise. The likelihood of our success must be considered in light of the problems, expenses, difficulties, complications, and delays frequently encountered in connection with the formation of a new business, the development of new technologies or those subject to clinical testing, and the competitive and regulatory environment in which we will operate. To date, we have generated revenue from a limited market launch of CyPath® Lung in the South Texas area which began in the second quarter of 2022. There can be no assurance that we will be able to successfully expand our commercialization efforts or that we will obtain the necessary regulatory approvals that will allow us to expand our marketing efforts. We may not be able to maintain certification of CyPath® Lung as an LDT in accordance with CAP/CLIA guidance and regulations, or obtain approval of our diagnostic tests in development by the CMS, the FDA, European Medicines Agency, or Chinese National Medical Products Administration. Even if we do so and are also able to commercialize our diagnostic tests, we may never generate revenue sufficient to become profitable. Our failure to generate revenue and profit would likely cause our securities to decrease in value or become worthless.
We will require additional financing to implement our Business Plan, which may not be available on favorable terms or at all, and we may have to accept financing terms that would place restrictions on us.
We believe that we must raise additional funds to be able to continue our business operations. We may not be able to obtain equity or debt financing on acceptable terms or at all to implement our growth strategy. As a result, adequate capital may not be available to finance our current development plan, take advantage of business opportunities, or respond to competitive pressures. If we are unable to raise additional funds, we may be forced to curtail or even abandon our Business Plan and focus on fewer commercial opportunities that may result in more limited growth than forecast.
Until such time, if ever, as we can generate substantial income from sale of our diagnostic test(s) and therapeutic product candidates, we expect to finance our cash needs through a combination of equity offerings, debt financings, and license and collaboration agreements. To the extent that we raise additional capital through the sale of equity or convertible debt securities, the ownership interest of existing stockholders will be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect the rights of the holders of our Common Stock (the “Common Stockholders”). In addition, the terms of any future financings may impose restrictions on our right to declare dividends or on the manner in which we conduct our business. Debt financing and preferred equity financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures, declaring dividends, or making acquisitions or significant asset sales.
If we raise additional funds through collaborations, strategic alliances or marketing, or distribution or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams, research programs; or grant licenses on terms that may not be favorable to us and/or that may reduce the value of our Common Stock.
Risks Related to our Diagnostic Product
Until we secure FDA clearance for our CyPath® Lung as a Class II in vitro diagnostic, our marketing efforts are limited.
In order to market our CyPath® Lung as an IVD medical device, we must receive de novo classification from the FDA as a Class II in vitro diagnostic. We intend to launch a pivotal trial later this year in an effort to attain such classification; however, there can be no assurance that the trial will have favorable results or that it will generate the results necessary to obtain such classification. Until such time as we receive de novo classification, which we may never receive, our marketing efforts are limited to the marketing and sale of CyPath® Lung as an LDT within those states and territories of the United States where PPLS is licensed or otherwise permitted under applicable law to offer, sell and market CyPath® Lung.
If we experience delays or difficulties in the enrollment of patients in clinical trials, our receipt of necessary regulatory approvals could be delayed or prevented.
We may not be able to initiate or continue clinical trials if we are unable to locate and enroll a sufficient number of eligible patients to participate in these trials as required by the FDA or similar regulatory authorities outside the United States, such as the European Medicines Agency.
Patient enrollment is affected by many other factors, including:
| ● | the severity of the disease under investigation; | |
| ● | the patient eligibility criteria for the study in question; | |
| ● | the efforts to facilitate timely enrollment in clinical trials; | |
| ● | our payments for conducting clinical trials; | |
| ● | the patient referral practices of physicians; | |
| ● | the ability to monitor patients adequately during the trial period; and | |
| ● | the proximity and availability of clinical trial sites for prospective patients. |
We are unable to forecast with precision our ability to enroll patients. Our inability to enroll a sufficient number of patients for our clinical trials would result in significant delays and could require us to abandon one or more clinical trials altogether. Enrollment delays in our clinical trials may result in increased development costs, which would cause the value of our Company to decline and limit our ability to obtain additional financing.
| 17 |
Clinical trials are expensive, time-consuming, and may not be successful.
Clinical trials are expensive, time-consuming, and may not be successful. They involve the evaluation of diagnostic tests and testing of potential therapeutic agents and effective treatments in humans to determine the safety and efficacy of the diagnostic tests and therapeutic products necessary for an approved diagnostic and therapeutic technology. Many tests and products in human clinical trials fail to demonstrate the desired safety and efficacy characteristics. Even if our tests and products progress successfully through initial or subsequent human testing, they may fail in later phases of development. We may engage others to conduct our clinical trials, including clinical research organizations and government-sponsored agencies. These trials may not start or be completed as we forecast or may not achieve desired results.
We may experience numerous unforeseen events during, or as a result of, clinical trials that could delay or prevent our ability to receive marketing authorization or commercialize our diagnostic and therapeutic technologies, including:
| ● | regulators or institutional review boards may not authorize us or our investigators to commence a clinical trial or conduct a clinical trial at a prospective trial site; | |
| ● | we may experience delays in reaching, or fail to reach, agreement on acceptable clinical trial contracts or clinical trial protocols with prospective trial sites; | |
| ● | clinical trials may produce negative or inconclusive results, and we may decide, or regulators may require us, to conduct additional clinical trials or abandon product and test development programs; | |
| ● | the number of patients required for clinical trials may be larger than we anticipate, enrollment in these clinical trials may be slower than we anticipate, or participants may drop out of these clinical trials at a higher rate than we anticipate; | |
| ● | our third-party contractors may fail to comply with regulatory requirements or meet their contractual obligations to us in a timely manner, or at all; | |
| ● | we may have to suspend or terminate clinical trials for various reasons, including a finding that the participants are being exposed to unacceptable health risks; | |
| ● | regulators or institutional review boards may require that we or our investigators suspend or terminate clinical research for various reasons, including noncompliance with regulatory requirements or a finding that the participants are being exposed to unacceptable health risks; | |
| ● | the cost of clinical trials may be greater than we anticipate; or | |
| ● | regulators may revise the requirements for approving our diagnostic or therapeutic technologies, or such requirements may not be as we anticipate. |
If we are required to conduct additional clinical trials or other testing beyond those that we currently contemplate, if we are unable to successfully complete clinical trials or other testing, if the results of these trials or tests are not positive or are only modestly positive, or if there are safety concerns, we may:
| ● | be delayed in obtaining marketing approval; | |
| ● | not obtain marketing approval at all, which would seriously impair our viability; | |
| ● | obtain marketing approval in some countries and not in others; | |
| ● | obtain approval for indications or patient populations that are not as broad as we intend or desire; | |
| ● | obtain approval with labeling that includes significant use or distribution restrictions or safety warnings; | |
| ● | be subject to additional post-marketing testing requirements; or | |
| ● | have the diagnostic test or therapeutic product removed from the market after obtaining marketing approval. |
Our product and test development costs will increase if we experience delays in clinical testing or marketing approvals. We do not know whether any of our preclinical studies or clinical trials will begin as planned, will need to be restructured, or will be completed on schedule or at all. Significant preclinical or clinical trial delays also could shorten any periods during which we may have the exclusive right to commercialize our diagnostic technology or allow our competitors to bring diagnostic tests and therapeutic products to market before we do, potentially impairing our ability to successfully commercialize our diagnostic and therapeutic technologies and harming our business and results of operations.
| 18 |
Risks Related to Our Diagnostic Tests
If our tests do not perform as expected, our operating results, reputation and business will suffer.
Our success depends on the market’s confidence that PPLS can provide reliable, high-quality clinical testing services. There is no guarantee that the accuracy and reproducibility that the CAP/CLIA clinical pathology laboratory, now owned by PPLS, has demonstrated to date will continue as its test volume increases. We believe that PPLS’ customers are likely to be particularly sensitive to test limitations and errors, including inaccurate test results. As a result, if PPLS does not perform its diagnostic services as expected, our operating results, reputation and business will suffer. We may be subject to legal claims arising from such limitations, errors or inaccuracies.
We may experience difficulties that delay or prevent our development, introduction or marketing of enhanced or new tests.
Our success may also depend on our ability to effectively introduce enhanced or new tests. The development of enhanced or new tests is complex, costly and uncertain. Furthermore, enhancing or developing new tests requires us to anticipate patients’, clinicians’ and payors’ needs and emerging technology trends accurately. We may experience research and development, regulatory, marketing and other difficulties that could delay or prevent our introduction of enhanced or new tests. The research and development process in diagnostics generally takes a significant amount of time from the research and design stage to commercialization. This process is conducted in various stages, and each stage presents the risk that we will not achieve our goals. We may have to abandon a test in which we have invested substantial resources. In order to successfully commercialize tests that we may develop in the future, we may need to conduct lengthy, expensive clinical trials and develop dedicated sales and marketing operations or enter into collaborative agreements to achieve market awareness and demand. Any delay in the research and development, approval, production, marketing or distribution of enhanced or new tests could adversely affect our competitive position, branding and results of operations.
We cannot be certain that:
| ● | any tests that we may enhance or develop will prove to be effective in clinical trials; |
| ● | we will be able to obtain, in a timely manner or at all, regulatory approvals, if needed; |
| ● | any tests that we may enhance or develop will be ordered and used by healthcare providers; |
| ● | any tests that we may enhance or develop can be provided at acceptable cost and with appropriate quality; or |
| ● | any of our tests can be successfully marketed. |
These factors and other factors beyond our control could delay the launch of enhanced or new tests.
If clinical testing of a particular diagnostic test or therapeutic product candidate does not yield successful results, then we will be unable to commercialize that test or product candidate.
We must demonstrate the product safety and efficacy of our candidates for diagnostic tests and therapeutic products in humans through extensive clinical testing. Our research and development programs are at an early stage of development. We may experience numerous unforeseen events during, or as a result of, the testing process that could delay or prevent commercialization of any test or product, including the following:
| ● | the results of pre-clinical studies may be inconclusive, or they may not be indicative of results that will be obtained in human clinical trials; | |
| ● | safety and efficacy results attained in early human clinical trials may not be indicative of results that are obtained in later clinical trials; |
| 19 |
| ● | after reviewing test results, we may abandon projects that we might previously have believed to be promising; | |
| ● | we or our regulators may suspend or terminate clinical trials because the participating subjects or patients are being exposed to unacceptable health risks; and | |
| ● | our test or product candidates may not have the desired effects or may include undesirable side effects or other characteristics that preclude regulatory approval or limit their commercial use if approved. |
Even if our diagnostic tests or therapeutic products receive marketing approval, they may fail to achieve the degree of market acceptance by physicians, patients, third-party payors and others in the medical community necessary for commercial success.
Even if our products receive marketing approval, if needed, they may nonetheless fail to gain sufficient market acceptance by physicians, patients, third-party payors, and others in the medical community. If we do not generate significant product revenues, we may not become profitable. The degree of market acceptance of our products and tests, if approved for commercial sale, will depend on a number of factors, including:
| ● | their efficacy, safety, and other potential advantages compared to alternative tests or products; | |
| ● | our ability to offer them for sale at competitive prices; | |
| ● | their convenience and ease of administration compared to alternative diagnostics or treatments; | |
| ● | the willingness of the target patient population to try new diagnostic tests and of physicians to order these tests; | |
| ● | the willingness of the target patient population to try new therapies and of physicians to prescribe these therapies; | |
| ● | the strength of marketing and distribution support; | |
| ● | the availability of governmental agencies and third-party medical insurance and adequate reimbursement for our diagnostic tests or therapeutic products; | |
| ● | any restrictions on the use of our diagnostic tests or therapeutic products together with other diagnostic methods or therapeutic treatments; | |
| ● | any restrictions on the use of our diagnostic tests or therapeutic products together with other medications; | |
| ● | inability of certain types of patients to produce adequate samples for analysis in the use of our diagnostic tests; | |
| ● | inability of certain types of patients to use our diagnostic tests or take our therapeutic products; and | |
| ● | the prevalence and severity of side effects from our therapeutic products. |
If we are unable to address and overcome these and similar concerns, our business and results of operations could be substantially harmed.
If we are unable to establish effective sales, marketing, and distribution capabilities or enter into agreements with third parties with such capabilities, we may not be successful in commercializing our diagnostic tests or therapeutic products if and when they are approved.
We do not have a sales or marketing infrastructure and have limited experience in the sale, marketing, or distribution of our diagnostic tests and therapeutic products. To achieve commercial success for any diagnostic test or therapeutic product for which we obtain marketing approval, we will need to successfully establish and maintain relationships directly and with third parties to perform sales and marketing functions.
Factors that may inhibit our efforts to commercialize our diagnostic tests or therapeutic products on our own include:
| ● | our inability to recruit, train, and retain adequate numbers of effective sales, technical support, and marketing personnel; | |
| ● | the inability of sales personnel to obtain access to or educate physicians on the benefits of our diagnostic tests or therapeutic products; | |
| ● | the lack of complementary diagnostic tests or therapeutic products to be offered by sales personnel, which may put us at a competitive disadvantage relative to companies with more extensive diagnostic tests or therapeutic product lines; | |
| ● | unforeseen costs and expenses associated with creating an independent sales, technical support, and marketing organization; and | |
| ● | the inability to obtain sufficient coverage and reimbursement from third-party payors and governmental agencies. |
If we do not establish sales, marketing, and distribution capabilities successfully, either on our own or in collaboration with third parties, we will not be successful in commercializing our diagnostic tests or therapeutic products.
We are currently dependent upon one pathology laboratory to offer and perform CyPath® Lung.
We previously granted Village Oaks a license pursuant to a joint development and project agreement to develop CyPath® Lung for commercialization and to manufacture, use, market and sell CyPath® Lung as an LDT within those states and territories of the United States where Village Oaks is licensed or otherwise permitted under applicable law to offer, sell and market CyPath® Lung, pursuant to which we received 50% of the gross revenue received by Village Oaks from the sale and processing of the CyPath® Lung test. In connection with the Acquisition, PPLS acquired the license to CyPath® Lung. Upon consolidating PPLS, our wholly owned subsidiary, the royalty revenue in connection with the performing of CyPath® Lung will be eliminated as an intercompany transaction. PPLS is currently the only licensee of CyPath® Lung and, therefore, we are dependent upon the efforts of PPLS, a CAP/CLIA clinical laboratory that performs CyPath® Lung, for the generation of our revenue. Revenue from CyPath Lung is generated through performance of testing by PPLS. PPLS performs testing when ordered by physicians for their patients. PPLS also generates revenue when performed in the context of an observational study conducted by the Department of Defense (the “DOD”) titled “Detection of Abnormal Respiratory Cell Populations in Lung Cancer Screening Patients Using the CyPath® Lung Assay,” and when performed for research and development on using bronchoalveolar lavage fluid as a biological sample to assess cardiopulmonary function and exercise performance in military personnel post COVID-19 infection.
| 20 |
If we are unable to convince physicians of the benefits of our proposed diagnostic tests or therapeutic products, we may incur delays or additional expense in our attempt to establish market acceptance.
Broad use of our proposed diagnostic tests and products may require pathology laboratories and physicians to be informed regarding our proposed diagnostic tests and products and their intended benefits. Inability to carry out this physician education process may adversely affect market acceptance of our proposed diagnostic tests or therapeutic products. We may be unable to timely educate physicians regarding our proposed diagnostic tests or therapeutic products in sufficient numbers to achieve our marketing plans or to achieve acceptance of our diagnostic tests or therapeutic products. Any delay in physician education may materially delay or reduce demand for our diagnostic tests or therapeutic products. In addition, we may expend significant funds toward physician education before any acceptance or demand for our proposed diagnostic tests or therapeutic products is created, if at all.
We face substantial competition, which may result in others discovering, developing, or commercializing competing diagnostic tests or therapeutic products before or more successfully than we do.
The development and commercialization of new diagnostic and therapeutic technologies is highly competitive. We will always face competition with respect to any diagnostic and therapeutic technology that we may seek to develop or commercialize in the future from major diagnostic and pharmaceutical companies, LDT laboratories, smaller diagnostic and pharmaceutical companies, and biotechnology companies worldwide. In 2022, we evaluated 67 companies advancing tests for the early detection of lung cancer that provided at least a scientific foundation for their tests. These competitors are investigating lung cancer screening and diagnostic methods that use various types of collected samples (blood, breath, nasal epithelial cells, saliva, sputum, and urine) or imaging systems. Potential competitors also include academic institutions, government agencies, and other public and private research organizations that conduct research, seek patent protection, and establish collaborative arrangements for research, development, manufacturing, and commercialization.
A substantial number of the companies against which we are competing or we may compete against in the future may have, significantly greater financial resources, established presence in the market, and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals, and marketing approved diagnostic tests or therapeutic products. Mergers and acquisitions in the diagnostic, pharmaceutical, and biotechnology industries may result in even more resources being concentrated among a smaller number of our competitors.
Smaller and other early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These third parties compete with us in recruiting and retaining qualified scientific, sales, marketing, and management personnel, establishing clinical trial sites and patient registration for clinical trials, and acquiring technologies complementary to or necessary for – our programs.
Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize diagnostic tests or therapeutic products that are more accurate, more convenient, or less expensive than any diagnostic tests or therapeutic products that we may develop. Our competitors also may obtain FDA or other regulatory approval for their diagnostic tests or therapeutic products more rapidly than we may obtain approval for ours, which could result in our competitors establishing a stronger market position. In addition, our ability to compete may be affected in many cases by insurers or other third-party payors.
We may be unable to compete in our target marketplaces, which could impair our ability to generate revenues, thus causing a material adverse impact on our results of operations.
Our success depends upon our ability to retain key executives and to attract, retain, and motivate qualified personnel, and the loss of these persons could adversely affect our operations and results.
We are highly dependent on the principal members of our management, scientific, and clinical teams, including Maria Zannes, J.D., our President and Chief Executive Officer, Vivienne Rebel, M.D., Ph.D., our Chief Science and Medical Officer and Executive Vice President, Xavier Reveles, MS, CG(ASCP)cm, our Chief Operating Officer, and Michael Dougherty, CPA, MBA, our Chief Financial Officer, as well as Roby Joyce, M.D., the Medical Director and Laboratory Director of PPLS and the principal of Village Oaks.
The loss of the services of any of our executive officers or other members of our management team could impede the achievement of our research, development, and commercialization objectives and seriously harm our ability to successfully implement our business strategy. Furthermore, replacing executive officers and key employees may be difficult and may take an extended period of time because of the limited number of individuals in our industry with the breadth of skills and experience required to successfully develop, gain regulatory approval of, and commercialize diagnostic tests or therapeutic products. Competition to hire from this limited pool is intense, and we may be unable to hire, train, retain, or motivate key personnel on acceptable terms given the competition among numerous biotechnology companies for similar expertise. We also face competition from universities and research institutions for qualified scientific and clinical personnel. In addition, we rely and expect to continue to rely to a significant degree on consultants and advisors, including scientific and clinical advisors, to assist us in formulating our research and development and commercialization strategies. Our consultants and advisors may be engaged by other entities and may have commitments under consulting or advisory contracts that may limit their availability to us. If we are unable to continue to attract and retain high-quality personnel, our ability to pursue our growth strategy will be limited.
Our lack of operating experience may make it difficult to manage our growth which could lead to our inability to implement our Business Plan.
We have limited experience in marketing and the selling of diagnostic tests and pharmaceutical products. Any growth will require us to expand our management and our operational and financial systems and controls. If we are unable to do so, our business and financial condition would be materially harmed. If rapid growth occurs, it may strain our operational, managerial, and financial resources.
| 21 |
If we fail to comply with our obligations imposed by any intellectual property licenses with third parties that we may need in the future, we could lose rights that are important to our business.
We may in the future require licenses to third-party technology and materials. We had previously been granted a license from Village Oaks to use its intellectual property, pursuant to a joint development and project agreement, to develop CyPath® Lung for commercialization. In connection with the Acquisition, Village Oaks assigned its rights pursuant to such joint development and project agreement to PPLS, as well as the intellectual property that is the subject of our license under such agreement. Such licenses may not be available in the future or may not be available on commercially reasonable terms, or at all, which could have a material adverse effect on our business and financial condition. We may rely on third parties from whom we license proprietary technology to file and prosecute patent applications and maintain patents and otherwise protect the intellectual property we license from them. We may have limited control over these activities or any other intellectual property that may be related to our in-licensed intellectual property. For example, we cannot be certain that such activities by these licensors will be conducted in compliance with applicable laws and regulations or will result in valid and enforceable patents and other intellectual property rights. We may have limited control over the manner in which our licensors initiate an infringement proceeding against a third-party infringer of the intellectual property rights or defend certain of the intellectual property that may be licensed to us. It is possible that the licensors’ infringement proceeding or defense activities may be less vigorous than if we conduct them ourselves. Even if we acquire the right to control the prosecution, maintenance, and enforcement of the licensed and sublicensed intellectual property relating to our diagnostic tests or therapeutic product candidates, we may require the cooperation of our licensors and any upstream licensor, which may not be forthcoming. Therefore, we cannot be certain that the prosecution, maintenance, and enforcement of these patent rights will be in a manner consistent with the best interests of our business. If we or our licensor fail to maintain such patents, or if we or our licensor lose rights to those patents or patent applications, the rights we have licensed may be reduced or eliminated and our right to develop and commercialize any of our diagnostic tests or therapeutic product candidates that are the subject of such licensed rights could be adversely affected. In addition to the foregoing, the risks associated with patent rights that we license from third parties will also apply to patent rights we may own in the future. Further, if we fail to comply with our diligence, development and commercialization timelines, milestone payments, royalties, insurance, and other obligations under our license agreements, we may lose our patent rights with respect to such agreement, which would affect our patent rights worldwide.
Termination of our current or any future license agreements would reduce or eliminate our rights under these agreements and may result in our having to negotiate new or reinstated agreements with less favorable terms or cause us to lose our rights under these agreements, including our rights to important intellectual property or technology. Any of the foregoing could prevent us from commercializing our other diagnostic tests or therapeutic product candidates, which could have a material adverse effect on our operating results and overall financial condition.
In addition, intellectual property rights that we in-license in the future may be sublicenses under intellectual property owned by third parties, in some cases through multiple tiers. The actions of our licensors may therefore affect our rights to use our sublicensed intellectual property, even if we are in compliance with all of the obligations under our license agreements. Should our licensors or any of the upstream licensors fail to comply with their obligations under the agreements pursuant to which they obtain the rights that are sublicensed to us, or should such agreements be terminated or amended, our ability to develop and commercialize our diagnostic tests or therapeutic product candidates may be materially harmed.
In the future, we may need to obtain additional licenses of third-party technology that may not be available to us or are available only on commercially unreasonable terms, which may cause us to operate our business in a more costly or otherwise adverse manner that was not anticipated.
We currently own intellectual property directed to our diagnostic tests, therapeutic product candidates and other proprietary technologies. Other pharmaceutical companies and academic institutions may also have filed or are planning to file patent applications potentially relevant to our business. From time to time, in order to avoid infringing these third-party patents, we may be required to license technology from additional third parties to further develop or commercialize our diagnostic tests or therapeutic product candidates. Should we be required to obtain licenses to any third-party technology, including any such patents required to manufacture, use, or sell our product candidates, such licenses may not be available to us on commercially reasonable terms, or at all. The inability to obtain any third-party license required to develop or commercialize any of our product candidates could cause us to abandon any related efforts, which could seriously harm our business and operations. The licensing or acquisition of third-party intellectual property rights is a competitive area, and several more established companies may pursue strategies to license or acquire third-party intellectual property rights we may consider attractive or necessary. These established companies may have a competitive advantage over us due to their size, capital resources, and greater clinical development and commercialization capabilities. In addition, companies that perceive us to be a competitor may be unwilling to assign or license rights to us. Even if we are able to obtain a license under such intellectual property rights, any such license may be non-exclusive, which may allow our competitors access to the same technologies licensed to us.
Moreover, some of our owned and in-licensed patents or patent applications or future patents may be co-owned with third parties. If we are unable to obtain an exclusive license to any such third-party co-owners’ interest in such patents or patent applications, such co-owners may be able to license their rights to other third parties, including our competitors, and our competitors could market competing diagnostic tests or therapeutic products and technology. In addition, we may need the cooperation of any such co-owners of our patents in order to enforce such patents against third parties, and such cooperation may not be provided to us. Furthermore, our owned and in-licensed patents may be subject to a reservation of rights by one or more third parties. Any of the foregoing could have a material adverse effect on our competitive position, business, financial conditions, results of operations and prospects.
We will depend on third parties to manufacture and market our diagnostic tests and to design trial protocols, arrange for and monitor the clinical trials, and collect and analyze data.
We do not have, and do not now intend to develop, facilities for the manufacture of the contents of our collection kits needed for clinical or commercial production. In addition, we are not a party to any long-term agreement with any of our suppliers such as the reagents used in processing sputum samples, and accordingly, we have the products used in our diagnostic tests manufactured on a purchase-order basis from primary suppliers. We have entered into relationships with manufacturers on a contract basis but will need to expand those relationships. We expect to depend on such collaborators to supply us with reagents and other materials manufactured in compliance with standards imposed by the CMS, FDA, and foreign regulators.
Moreover, as we develop our diagnostic tests or therapeutic products eligible for clinical trials, we intend to contract with independent parties to design the trial protocols, arrange for and monitor the clinical trials, and collect and analyze the data. In addition, certain clinical trials for our products may be conducted by government-sponsored agencies and will be dependent on governmental participation and funding. Our dependence on independent parties and clinical sites involves risks including reduced control over the timing and other aspects of our clinical trials.
| 22 |
We are exposed to product liability and pre-clinical and clinical liability risks which could place a substantial financial burden upon us, should we be sued.
Our business exposes us to potential product liability and other liability risks that are inherent in the testing, manufacturing, and marketing of diagnostic tests and therapeutic products. Such claims may be asserted against us. In addition, using diagnostic tests and therapeutic products that may be developed with potential collaborators in our clinical trials and the subsequent sale of these tests and products by bioAffinity or our potential collaborators may cause us to bear a portion of or all product liability risks. A successful liability claim, or series of claims, brought against us could have a material adverse effect on our business, financial condition, and results of operations.
While we have obtained product liability insurance covering CyPath® Lung as a commercialized LDT to be sold by a CAP-accredited, CLIA-certified clinical pathology laboratory (previously Village Oaks and currently PPLS), in the future we may not be able to obtain or maintain adequate product liability insurance, when needed, on acceptable terms, if at all, or such insurance may not provide adequate coverage against our potential liabilities. Furthermore, potential partners with whom we intend to have collaborative or strategic agreements or our future licensees may not be willing to indemnify us against these types of liabilities and may not themselves be sufficiently insured or have sufficient liquidity to satisfy any product liability claims. Claims or losses in excess of any product liability insurance coverage that we may obtain could have a material adverse effect on our business, financial condition, and results of operations.
In addition, we may be unable to obtain or to maintain clinical trial liability insurance on acceptable terms, if at all. Any inability to obtain and/or maintain insurance coverage on acceptable terms could prevent or limit the commercialization of any tests or products we develop.
Our collection, use and disclosure of personal information, including health and employee information, is subject to U.S. state and federal privacy and security regulations, and our failure to comply with those regulations or to adequately secure the information we hold could result in significant liability or reputational harm.
The privacy and security of personal information stored, maintained, received or transmitted, including electronically, is a major issue in the U.S. and abroad. Numerous federal and state laws and regulations, including state privacy, data security and breach notification laws, federal and state consumer protection and employment laws, the Health Insurance Portability and Accountability Act of 1996, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009, and the Genetic Information Nondiscrimination Act of 2008, govern the collection, dissemination, use and confidentiality of personal information, including genetic, biometric and health information. These laws and regulations are increasing in complexity and number, may change frequently and sometimes conflict. Penalties for violations of these laws vary but can be severe.
While we strive to comply with all applicable privacy and security laws and regulations, including our own posted privacy policies, these laws and regulations continue to evolve, and any failure or perceived failure to comply may result in proceedings or actions against us by government entities or others or could cause us to lose customers, which could have a material adverse effect on our business. Recently, there has been an increase in public awareness of privacy issues in the wake of revelations about the data-collection activities of various government agencies and in the number of private privacy-related lawsuits filed against companies. Concerns about our practices with regard to the collection, use, retention, disclosure, or security of personal information or other privacy-related matters, even if unfounded and even if we are in compliance with applicable laws, could damage our reputation and harm our business.
If users of our proposed diagnostic tests or therapeutic products are unable to obtain adequate reimbursement from third-party payors or governmental agencies or if new restrictive legislation is adopted, market acceptance of our proposed tests or products may be limited, and we may not achieve revenues.
The continuing efforts of government and insurance companies, health maintenance organizations (“HMOs”) and other payors of healthcare costs to contain or reduce costs may affect our future revenues and profitability, as well as the future revenues and profitability of our potential customers, suppliers, and collaborative partners and the availability of capital. For example, in certain international markets, pricing or profitability of diagnostic tests and therapeutic products is subject to government control. In the U.S., given recent federal and state government initiatives directed at lowering the total cost of healthcare, the U.S. Congress and state legislatures will likely continue to focus on healthcare reform, the cost of medical devices, tests, and prescription pharmaceuticals, and Medicare and Medicaid reforms. While we cannot predict whether any such legislative or regulatory proposals will be adopted, the announcement or adoption of such proposals could materially harm our business, financial condition, and results of operations.
Our ability to commercialize our proposed tests or products will depend in part on the extent to which appropriate reimbursement levels for the cost of our tests or products are obtained by governmental authorities, private health insurers, and other organizations such as HMOs. Governmental agencies and third-party payors are increasingly challenging the prices charged for medical tests, drugs, and services. Also, the trend toward managed healthcare in the U.S. and the concurrent growth of organizations such as HMOs, which could control or significantly influence the purchase of healthcare services, diagnostics, and drugs, as well as legislative proposals to reform healthcare or reduce government insurance programs, may all result in lower prices for or rejection of our tests or products.
Our employees, independent contractors, consultants, commercial partners, and vendors may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements.
Our business operations and current and future relationships with investigators, healthcare professionals, consultants, third-party payors and customers will be subject, directly or indirectly, to federal and state healthcare fraud and abuse laws, false claims laws, health information privacy and security laws, and other healthcare laws and regulations. If we are unable to comply, or have not fully complied, with such laws, we could face substantial penalties. We are exposed to the risk of employee fraud or other illegal activity by our employees, independent contractors, consultants, commercial partners, vendors and agents acting on behalf of us or our affiliates. Misconduct by these parties could include intentional, reckless and/or negligent conduct that fails to: comply with the regulations of the FDA or foreign health authorities; provide true, complete and accurate information to the FDA or foreign health authorities; comply with manufacturing standards we have established; comply with healthcare fraud and abuse laws in the U.S. and similar foreign fraudulent misconduct laws; or report financial information or data accurately or to disclose unauthorized activities to us.
| 23 |
Our business operations and current and future relationships with investigators, healthcare professionals, consultants, third-party payors and customers are subject, directly or indirectly, to federal and state healthcare fraud and abuse laws, transparency laws and other healthcare laws and regulations. If we are unable to comply, or have not fully complied, with such laws, we could face substantial penalties.
Healthcare providers and others play a primary role in the recommendation ordering and prescription of any diagnostic tests or therapeutic products for which we obtain marketing approval. Our operations and current and future arrangements with investigators, healthcare professionals, customers, and third-party payors are subject to various U.S. federal and state healthcare laws and regulations, including, without limitation, U.S. federal Anti-Kickback Statute, the U.S. federal civil and criminal false claims laws, and the Physician Payments Sunshine Act and regulations. These laws may impact, among other things, our current business operations, including our clinical research activities, and proposed sales, marketing, and education programs and constrain the business of financial arrangements and relationships with healthcare providers and other parties through which we may market, sell, and distribute our diagnostic tests or therapeutic products for which we obtain marketing approval. In addition, we may be subject to additional healthcare, statutory, and regulatory requirements and enforcement by foreign regulatory authorities in jurisdictions in which we conduct our business.
Ensuring that our internal operations and future business arrangements with third parties comply with applicable healthcare laws and regulations will involve substantial costs. It is possible that governmental authorities will conclude that our business practices, including certain arrangements with physicians who receive stock, warrants or stock options as compensation for services provided to us, do not comply with current or future statutes, regulations, agency guidance, or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of the laws described above or any other governmental laws and regulations that may apply to us, we may be subject to significant penalties, including civil, criminal and administrative penalties, damages, fines, exclusion from U.S. government-funded healthcare programs, such as Medicare and Medicaid, or similar programs in other countries or jurisdictions, disgorgement, imprisonment, contractual damages, reputational harm, diminished profits, additional reporting requirements and oversight if we become subject to a corporate integrity agreement or similar agreement to resolve allegations of non-compliance with these laws, and the delay, reduction, termination or restructuring of our operations. Further, defending against any such actions can be costly and time-consuming and may require significant financial and personnel resources. Therefore, even if we are successful in defending against any such actions that may be brought against us, our business may be impaired. If any of the physicians or other providers or entities with whom we expect to do business are found not to be in compliance with applicable laws, they may be subject to significant criminal, civil or administrative sanctions, including exclusions from government-funded healthcare programs and imprisonment. If any of the above occur, it could adversely affect our ability to operate our business and our results of operations.
We face intense competition in the biotechnology and pharmaceutical industries.
The biotechnology and pharmaceutical industries are intensely competitive. We face direct competition from U.S. and foreign companies focusing on diagnostic tests and pharmaceutical products, which are rapidly evolving. Our competitors include major multinational diagnostic and pharmaceutical companies, specialized biotechnology firms, and universities and other research institutions. Many of these competitors have greater financial and other resources, larger research and development staffs, and more effective marketing and manufacturing organizations than we do. In addition, academic and government institutions are increasingly likely to enter into exclusive licensing agreements with commercial enterprises, including our competitors, to market commercial tests or products based on technology developed at such institutions. Our competitors may succeed in developing or licensing technologies, tests, and products that are more effective or less costly than ours or succeed in obtaining CAP/CLIA validation or FDA or other regulatory approvals for diagnostic test and therapeutic product candidates before we do. Acquisitions of, or investments in, competing diagnostic, pharmaceutical, or biotechnology companies by large corporations could increase such competitors’ financial, marketing, manufacturing, and other resources.
The market for our proposed tests and products is competitive and rapidly changing, and new diagnostic technologies which may be developed by others could impair our ability to maintain and grow our business and remain competitive.
The diagnostic, pharmaceutical, and biotechnology industries are subject to rapid and substantial technological change. Developments by others may render our proposed tests or products noncompetitive or obsolete, or we may be unable to keep pace with technological developments or other market factors. Technological competition from diagnostic, pharmaceutical and biotechnology companies, universities, governmental entities, and others diversifying into the field is intense and is expected to increase.
As a company engaged in the development of diagnostic technology with limited revenue generated to date, our resources are limited, and we may experience technical challenges inherent in such technologies. Competitors have developed or are in the process of developing technologies that are, or in the future may be, the basis for competition. Some of these technologies may have an entirely different approach or means of accomplishing similar diagnostic efficacy compared to our proposed tests or products. Our competitors may develop diagnostic technologies that are more effective or less costly than our proposed tests or products and therefore present a serious competitive threat.
The potential widespread acceptance of diagnostic tests or therapies that are alternatives to ours may limit market acceptance of our proposed tests or products, even if commercialized. Many of our targeted diseases and conditions can also be detected by other tests or treated by other medications. These tests and treatments may be widely accepted in medical communities and have a longer history of use. The established use of these competitive technologies may limit the potential for our technologies, formulations, tests, and products to receive widespread acceptance if commercialized.
Healthcare cost containment initiatives and the growth of managed care may limit our returns.
Our ability to commercialize our diagnostic tests and therapeutic products successfully may be affected by the ongoing efforts of governmental and third-party payors to contain the cost of healthcare. These entities are challenging prices of healthcare products and services, denying or limiting coverage and reimbursement amounts for new diagnostic tests and therapeutic products, CAP/CLIA-validated LDTs and FDA-approved diagnostic tests and therapeutic products considered experimental or investigational or which are used for disease indications without FDA marketing authorization. Even if we succeed in bringing any tests or products to the market, they may not be considered cost-effective, and governmental or third-party reimbursement might not be available or sufficient. If adequate governmental or third-party coverage is not available, we may not be able to maintain price levels sufficient to realize an appropriate return on our investment in research and development for new tests and products. In addition, legislation and regulations affecting the pricing of diagnostic tests, pharmaceuticals, or healthcare services may change in ways adverse to us before or after any of our proposed tests and products are approved for marketing.
| 24 |
Our competitive position depends on protection of our intellectual property.
Development and protection of our intellectual property are critical to our business. If we do not adequately protect our intellectual property, or if competitors develop technologies incorporating the same or similar technologies that already are in the public domain, those competitors may be able to develop similar technologies to our own. Our success depends in part on our ability to obtain patent protection for our diagnostic tests, therapeutic products, or processes in the U.S. and other countries, protect trade secrets, and prevent others from infringing on our proprietary rights.
Since patent applications in the U.S. are maintained in secrecy for at least portions of their pendency periods (published on U.S. patent issuance or, if earlier, 18 months from earliest filing date for most applications) and since other publication of discoveries in the scientific or patent literature often lags behind actual discoveries, we cannot be certain that we are or will be the first to make the inventions to be covered by our patent applications. The patent position of biopharmaceutical and biotechnology firms generally is highly uncertain and involves complex legal and factual questions. The U.S. Patent and Trademark Office has not established a consistent policy regarding the breadth of claims that it will allow in biotechnology patents.
The patent applications we file, including applications that will follow the filing of provisional patents, may not issue as patents or the claims of any issued patents may not afford meaningful protection for our technologies, tests, or products. In addition, patents issued to us or to any future licensors may be challenged and subsequently narrowed, invalidated, or circumvented. Patent litigation is widespread in the biotechnology industry and could harm our business. Litigation might be necessary to protect our patent position or to determine the scope and validity of third-party proprietary rights, and we may not have the required resources to pursue such litigation or to protect our patent rights.
Although we have executed assignment of invention agreements with current scientific and technical employees and in the future will require our scientific and technical employees and consultants to enter into broad assignment of invention agreements, and all of our employees, consultants, and corporate partners with access to proprietary information enter into confidentiality agreements, these agreements may not be honored.
Diagnostic tests and therapeutic products we develop could be subject to infringement claims asserted by others.
We cannot assure that diagnostic tests and therapeutic products based on our patents or intellectual property that we license from others will not be challenged by a third-party claiming infringement of its proprietary rights. If we are not able to successfully defend patents that may be issued to us, that we may acquire, or that we may license in the future, we may have to pay substantial damages or licensing fees, possibly including treble damages, for past infringement.
We may become involved in lawsuits to protect or enforce our patents or other intellectual property, which could be expensive, time-consuming, and ultimately unsuccessful.
Competitors may infringe our issued patents or other intellectual property. To counter infringement or unauthorized use, we intend to file infringement claims, which can be expensive and time-consuming. Any claims we assert against perceived infringers could provoke these parties to assert counterclaims against us alleging that we infringe their intellectual property. In addition, in a patent infringement proceeding, a court may decide that a patent of ours is invalid or unenforceable, in whole or in part, construe the patent’s claims narrowly, or refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover the technology in question. An adverse result in any litigation proceeding could put one or more of our patents at risk of being invalidated or interpreted narrowly, which could adversely affect us.
If we are unable to protect the confidentiality of our trade secrets, our business and competitive position would be harmed.
In addition to seeking patents for some of our technology, we also intend to rely on trade secrets, including unpatented know-how, technology, and other proprietary information, to maintain our competitive position. We have executed and will continue to seek to protect these trade secrets, in part, by entering into non-disclosure and confidentiality agreements with parties who have access to them, such as our employees, corporate collaborators, outside scientific collaborators, contract manufacturers, consultants, advisors, and other third parties. We also have executed and will continue to seek to enter into confidentiality and invention or patent assignment agreements with our employees and consultants. Despite these efforts, any of these parties may breach the agreements and disclose our proprietary information, including our trade secrets, and we may not be able to obtain adequate remedies for such breaches. Our trade secrets may also be obtained by third parties by other means, such as breaches of our physical or computer security systems.
Enforcing a claim that a party illegally disclosed or misappropriated a trade secret is difficult, expensive, and time-consuming, and the outcome is unpredictable. In addition, some courts inside and outside the U.S. are less willing or unwilling to protect trade secrets. If any of our trade secrets were to be lawfully obtained or independently developed by a competitor, we would have no right to prevent them, or those to whom they communicate it, from using that technology or information to compete with us. If any of our trade secrets were to be disclosed to or independently developed by a competitor, our competitive position would be harmed.
Our internal information technology systems, or those of our third-party clinical research organizations or other contractors or consultants, may fail or suffer security breaches, loss or leakage of data, and other disruptions, which could result in a material disruption of our diagnostic tests’ or therapeutic product candidates’ development programs, compromise sensitive information related to our business or prevent us from accessing critical information, potentially exposing us to liability or otherwise adversely affecting our business.
We are increasingly dependent upon information technology systems, infrastructure and data to operate our business. In the ordinary course of business, we collect, store and transmit confidential information (including but not limited to intellectual property, proprietary business information and personal information). It is critical that we do so in a secure manner to maintain the confidentiality and integrity of such confidential information. We have also outsourced elements of our operations to third parties, and as a result we manage a number of third-party contractors who have access to our confidential information.
| 25 |
Despite the implementation of security measures, given their size and complexity and the increasing amounts of confidential information that they maintain, our internal information technology systems and those of our third-party clinical research organizations and other contractors and consultants are potentially vulnerable to breakdown or other damage or interruption from service interruptions, system malfunction, natural disasters, terrorism, war and telecommunication and electrical failures, as well as security breaches from inadvertent or intentional actions by our employees, contractors, consultants, business partners, and/or other third parties, or from cyber-attacks by malicious third parties (including the deployment of harmful malware, ransomware, extortion, account takeover attacks, degradation of service attacks, denial-of-service attacks, “phishing,” or social engineering and other means to affect service reliability and threaten the confidentiality, integrity and availability of information), which may compromise our system infrastructure or lead to data leakage. We have technology security initiatives and disaster recovery plans in place to mitigate our risk to these vulnerabilities, but these measures may not be adequately designed or implemented to ensure that our operations are not disrupted or that data security breaches do not occur. To the extent that any disruption or security breach were to result in a loss of, or damage to, our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liability and reputational damage.
Hackers and data thieves are increasingly sophisticated and operate large-scale and complex automated attacks which may remain undetected until after they occur. We cannot assure you that our data protection efforts and our investment in information technology will prevent significant breakdowns, data leakages, breaches in our systems or other cyber incidents that could have a material adverse effect upon our reputation, business, operations or financial condition. For example, if such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our programs and the development of our diagnostic tests and therapeutic product candidates could be delayed. In addition, the loss of clinical trial data for our diagnostic tests and therapeutic product candidates could result in delays in our marketing approval efforts and significantly increase our costs to recover or reproduce the data. Furthermore, significant disruptions of our internal information technology systems or security breaches could result in the loss, misappropriation, and/or unauthorized access, use, or disclosure of, or the prevention of access to, confidential information (including trade secrets or other intellectual property, proprietary business information, and personal information), which could result in financial, legal, business, and reputational harm to us. Like all businesses we may be increasingly subject to ransomware or other malware that could significantly disrupt our business operations, or disable or interfere with necessary access to essential data or processes. Numerous recent attacks of this nature have also involved exfiltration and disclosure of sensitive or confidential personal or proprietary information, or intellectual property, when victim companies have not paid the cyber criminals substantial ransom payments. For example, any such event that leads to unauthorized access, use, disclosure, unavailability, or compromised integrity of personal or other sensitive or essential information, including personal information regarding our clinical trial subjects or employees, could harm our reputation directly, compel us to comply with federal and/or state breach notification laws and foreign law equivalents, subject us to mandatory corrective action, increase the costs we incur to protect against such information security breaches, such as increased investment in technology, render key personnel unable to perform duties or communicate throughout the organization and otherwise subject us to fines and other liability under laws and regulations that protect the privacy and security of personal information, which could result in significant legal and financial exposure and reputational damages that could potentially have an adverse effect on our business.
The costs of mitigating cybersecurity risks are significant and are likely to increase in the future. These costs include, but are not limited to, retaining the services of cybersecurity providers; compliance costs arising out of existing and future cybersecurity, data protection and privacy laws and regulations; and costs related to maintaining redundant networks, data backups and other damage-mitigation measures. We also cannot be certain that our existing insurance coverage will continue to be available on acceptable terms or in amounts sufficient to cover the potentially significant losses that may result from a security incident or breach or that the insurer will not deny coverage of any future claim.
Risks Related to the Operation of a CAP/CLIA Laboratory
The operations of PPLS will depend in part upon Dr. Roby Joyce and his relationship with existing customers and our ability to establish relationships with these customers.
PPLS’ future success will depend in significant part upon the continued relationships with existing customers, many of whom have developed professional relationships with Dr. Roby Joyce. While Dr. Joyce will be the Medical Director and Laboratory Director of PPLS and a member of the bioAffinity board of directors, we cannot assure you that we will be able to retain his services. Although we have entered into a three-year employment agreement with him, there can be no assurance that the agreement will not be terminated prior to its expiration. We do not have an insurance policy on the life of Dr. Joyce, and we do not have “key person” life insurance policies for any of our other officers or advisors. The loss of the technical knowledge and management and industry expertise of Dr. Joyce or any of our key personnel could result in delays in services, loss of customers and sales and diversion of management resources, which could adversely affect our operating results.
| 26 |
PPLS may be unable to effectively maintain PPLS’ equipment or generate revenue when its equipment is not operational.
Timely, effective service is essential to maintaining the reputation and high use rates of the CAP/CLIA laboratory now owned by PPLS. Although it has agreements with a third-party equipment service providers pursuant to which such service providers maintain and repair its equipment, the agreement does not compensate it for loss of revenue when its systems are not fully operational and its business interruption insurance may not provide sufficient coverage for the loss of revenue. Also, third-party equipment service providers may not be able to perform repairs or supply needed parts in a timely manner, which could result in a loss of revenue. Therefore, if PPLS experiences more equipment malfunctions than anticipated or if it is unable to promptly obtain the service necessary to keep its equipment functioning effectively, or where its business or data is compromised on account of equipment malfunctions or a cybersecurity-related attack, PPLS’s ability to provide services and to fulfill its contractual arrangements would be adversely affected and our revenue could decline.
If our sole laboratory facility becomes damaged or inoperable, loses its accreditation or is required to vacate the facility, PPLS’ ability to sell its products or provide diagnostic assays and pursue its research and development efforts may be jeopardized.
Our only CLIA-certified, CAP-accredited, and state-licensed laboratory was recently acquired from Village Oaks by our wholly owned subsidiary, PPLS. PPLS’ facilities and equipment could be harmed or rendered inoperable by natural or man-made disasters, including fire, earthquake, flooding and power outages, which may render it difficult or impossible for it to provide pathology services or perform our diagnostic assays for some period of time. The inability to of PPLS to perform its services for its customers if PPLS’ facility is inoperable for even a short period of time, may result in the loss of customers or harm to its reputation or relationships with its customers, and it may be unable to regain those customers or repair its reputation in the future. Furthermore, PPLS’ facilities and the equipment it uses to perform its services could be costly and time-consuming to repair or replace.
Further, if PPLS’ current or future CLIA-certified, CAP-accredited, and state-licensed laboratory becomes inoperable or unqualified in any way it may not be able to license or transfer its technology to another facility with the necessary qualifications, including state licensure and CLIA certification, under the scope of which its current assays and its planned future assays could be performed. Even if PPLS finds a facility with such qualifications to perform its assays, it may not be available to PPLS on commercially reasonable terms.
PPLS relies on commercial courier delivery services to transport sputum samples for processing the CyPath® Lung test in a timely and cost-efficient manner and if these delivery services are disrupted, its business will be harmed.
PPLS’ business depends on its ability to quickly and reliably deliver test results to its customers. Sputum samples are received overnight within the United States for analysis at the laboratory facility located in San Antonio, Texas. Disruptions in delivery service, whether due to bad weather, natural disaster, terrorist acts or threats or for other reasons could adversely affect specimen integrity and its ability to process samples in a timely manner and to service its customers, and ultimately its reputation and its business. In addition, if PPLS is unable to continue to obtain expedited delivery services on commercially reasonable terms, its operating results may be adversely affected.
Security breaches, loss of data and other disruptions could compromise sensitive information related to PPLS’ business or prevent it from accessing critical information and expose it to liability, which could adversely affect its, and our, business and reputation.
In the ordinary course of its business, PPLS collects and stores sensitive data, including legally-protected health information, credit card information and personally identifiable information, such as data collected in connection with the CyPath® Lung laboratory test results. PPLS also stores sensitive intellectual property and other proprietary business information, including that of its customers, payors and collaboration partners. PPLS manages and maintains its applications and data utilizing a combination of on-site systems, managed data center systems and cloud-based data center systems. These applications and data encompass a wide variety of business-critical information, including research and development information, commercial information and business and financial information. PPLS is highly dependent on information technology networks and systems, including the Internet, to securely process, transmit and store this critical information. Although its policies and practices adhere to the requirements of the Health Insurance Portability and Accountability Act of 1996 (“HIPAA”) and PPLS employs measures to protect sensitive information from unauthorized access or disclosure, its information technology and infrastructure, and that of its third-party billing and collections provider, may be vulnerable to attacks by hackers or viruses or breached due to employee error, malfeasance or other disruptions.
| 27 |
A security breach or privacy violation that leads to disclosure or modification of or prevents access to patient information, including personally identifiable information or protected health information, could harm PPLS’ reputation, compel PPLS to comply with state breach notification laws, subject PPLS to mandatory corrective action, require PPLS to verify the correctness of database contents and otherwise subject PPLS to liability under laws that protect personal data, resulting in increased costs or loss of revenue. If PPLS is unable to prevent such security breaches or privacy violations or implement satisfactory remedial measures, its operations could be disrupted, and it may suffer loss of reputation, financial loss and other regulatory penalties because of lost or misappropriated information, including sensitive patient data. In addition, these breaches and other inappropriate access can be difficult to detect, and any delay in identifying them may lead to increased harm of the type described above.
Any such breach or interruption could compromise PPLS’ networks, and the information stored there could be inaccessible or could be accessed by unauthorized parties, publicly disclosed, lost or stolen. Any such interruption in access, improper access, disclosure, modification of, or other loss of information could result in legal claims or proceedings, liability under laws that protect the privacy of personal information, such as HIPAA, and regulatory penalties. Unauthorized access, loss or dissemination could also disrupt PPLS’ operations, including its ability to perform tests, provide test results, bill payors or patients, process claims and appeals, provide customer assistance services, conduct research and development activities, develop and commercialize tests, collect, process and prepare company financial information, provide information about tests, educate patients and clinicians about services and manage the administrative aspects of its business, any of which could damage its, and our, reputation and adversely affect our business. Any such breach could also result in the compromise of PPLS’ trade secrets and other proprietary information, which could adversely affect our competitive position.
In addition, the interpretation and application of health-related, privacy and data protection laws in the United States, Europe and elsewhere are often uncertain, contradictory and in flux. It is possible that these laws may be interpreted and applied in a manner that is inconsistent with PPLS’ practices. If so, this could result in government-imposed fines or orders requiring that it change its practices, which could adversely affect our business and its, and our, reputation. Complying with these various laws could cause us to incur substantial costs or require PPLS to change its business practices and compliance procedures in a manner adverse to our business.
If PPLS uses hazardous chemicals in a manner that causes injury, PPLS could be liable for damages.
PPLS’ activities currently require the controlled use of potentially harmful chemicals. PPLS cannot eliminate the risk of accidental contamination or injury to employees or third parties from the use, storage, handling or disposal of these materials. In the event of contamination or injury, PPLS could be held liable for any resulting damages, and any liability could exceed its resources or any applicable insurance coverage it may have. Additionally, PPLS is subject to, on an ongoing basis, federal, state and local laws and regulations governing the use, storage, handling and disposal of these materials and specified waste products. The cost of compliance with these laws and regulations may become significant and could have a material adverse effect on its, and therefore our, financial condition, results of operations and cash flows. In the event of an accident or if PPLS otherwise fails to comply with applicable regulations, it could lose its permits or approvals or be held liable for damages or penalized with fines.
If PPLS are unable to successfully scale its operations to support demand for CyPath® Lung, its business could suffer.
As test volume of CyPath® Lung grows, PPLS will need to continue to ramp up its testing capacity, implement increases in scale and related processing, customer service, billing and systems process improvements, and expand its internal quality assurance program and technology platform to support testing on a larger scale. PPLS will also need additional equipment and certified laboratory personnel to process higher volumes of our tests. We cannot assure you that any increases in scale, related improvements and quality assurance will be successfully implemented by PPLS or that equipment and appropriate personnel will be available. As additional tests are developed, PPLS may need to bring new equipment on-line, implement new systems, technology, controls and procedures and hire personnel with different qualifications.
The value of CyPath® Lung depends, in large part, on PPLS’ ability to perform the tests on a timely basis and at high-quality, and on its reputation for such timeliness and quality. Failure to implement necessary procedures or to hire the necessary personnel could impact its ability to meet market demand. There can be no assurance that it will be able to perform tests on a timely basis at a level consistent with demand, that its efforts to scale its commercial operations will not negatively affect the quality of test results or that it will be successful in responding to the growing complexity of testing operations.
In addition, PPLS’ growth may place a significant strain on its management, operating and financial systems and its sales, marketing and administrative resources. As a result of its growth, PPLS’ operating costs may escalate even faster than planned, and some of its internal systems may need to be enhanced or replaced. If we cannot effectively manage PPLS’ expanding operations and its costs, we may not be able to grow effectively or we may grow at a slower pace, and our business could be adversely affected.
| 28 |
Billing for PPLS’ services is complex, and PPLS must dedicate substantial time and resources to the billing process to be paid.
Billing for clinical laboratory services is complex, time-consuming and expensive. Depending on the billing arrangement and applicable law, PPLS bills various payors, including Medicare, insurance companies and patients, all of which have different billing requirements. It generally bills third-party payors for its diagnostic assays and pursues reimbursement on a case-by-case basis where pricing contracts or Medicare reimbursement is not in place. To the extent laws or contracts require it to bill patient co-payments or co-insurance, PPLS must also comply with these requirements. PPLS may also face increased risk in its collection efforts, including potential write-offs of doubtful accounts and long collection cycles, which could adversely affect its business, results of operations and financial condition.
Several factors make the billing process complex, including:
| ● | the reimbursement rates of payors; | |
| ● | compliance with complex federal and state regulations related to billing Medicare; | |
| ● | risk of government audits related to billing Medicare; | |
| ● | disputes among payors as to which party is responsible for payment; | |
| ● | differences in coverage and in information and billing requirements among payors, including the need for prior authorization and/or advanced notification; | |
| ● | the effect of patient co-payments or co-insurance; | |
| ● | changes to billing codes and/or coverage policies that apply to PPLS’ assays; | |
| ● | incorrect or missing billing information; and | |
| ● | the resources required to manage the billing and claims appeals process. |
PPLS uses standard industry billing codes, known as Current Procedural Terminology, or CPT, codes, to bill for its diagnostic assays. These codes can change over time. When codes change, there is a risk of an error being made in the claim adjudication process. These errors can occur with claims submission, third-party transmission or in the processing of the claim by the payor. Claim adjudication errors may result in a delay in payment processing or a reduction in the amount of the payment received. Coding changes, therefore, may have an adverse effect on PPLS’ revenues. There can be no assurance that payors will recognize these codes in a timely manner or that the process of transitioning to such a code and updating their billing systems will not result in errors, delays in payments and a related increase in accounts receivable balances.
As PPLS introduces new assays, PPLS will need to add new codes to its billing process as well as its financial reporting systems. Failure or delays in effecting these changes in external billing and internal systems and processes could negatively affect its collection rates, revenue, and cost of collecting.
Additionally, PPLS’ billing activities require its third-party billing provider to implement compliance procedures and oversight, train and monitor its employees, challenge coverage and payment denials, assist patients in appealing claims, and require PPLS to undertake audits to evaluate compliance with applicable laws and regulations as well as internal compliance policies and procedures. Payors also conduct external audits to evaluate payments, which add further complexity to the billing process. If the payor makes an overpayment determination, there is a risk that PPLS may be required to return some portion of prior payments it has received. These billing complexities and the related uncertainty in obtaining payment for its assays could negatively affect its revenue and cash flow, its ability to achieve profitability, and the consistency and comparability of its, and therefore our, results of operations.
PPLS relies on a third-party billing provider and an in-house billing function to transmit claims to payors, and any delay in transmitting claims could have an adverse effect on its revenue.
While PPLS manages the overall processing of claims, it relies on a third-party billing provider to transmit the actual claims to payors based on the specific payor billing format. Claims processing could be delayed if its third-party provider makes changes to its invoicing system. Additionally, coding for diagnostic assays may change, and such changes may cause short-term billing errors that may take significant time to resolve. If claims are not submitted to payors on a timely basis or are erroneously submitted, or if PPLS is required to switch to a different provider to handle claim submissions, it may experience delays in its ability to process these claims and receipt of payments from payors, or possibly denial of claims for lack of timely submission, which would have an adverse effect on its, and therefore our, revenue and business.
| 29 |
Risks Related to Intellectual Property Rights
Intellectual property rights do not necessarily address all potential threats to our competitive advantage.
The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations and may not adequately protect our business or permit us to maintain our competitive advantage. For example:
| ● | others may be able to make diagnostic tests and therapeutic product candidates that are the same as or similar to ours but that are not covered by the claims of the patents that we own or have exclusively licensed; | |
| ● | we or our licensors or future collaborators might not have been the first to make the inventions covered by the issued patent or pending patent application that we own or have exclusively licensed; | |
| ● | we or our licensors or future collaborators might not have been the first to file patent applications covering certain of our inventions; | |
| ● | others may independently develop similar or alternative technologies or duplicate any of our technologies without infringing our intellectual property rights; | |
| ● | it is possible that noncompliance with the U.S. Patent and Trademark Office (“USPTO”) and foreign governmental patent agencies requirement for a number of procedural, documentary, fee payment and other provisions during the patent process can result in abandonment or lapse of a patent or patent application, and partial or complete loss of patent rights in the relevant jurisdiction; | |
| ● | it is possible that our pending patent applications will not lead to issued patents; | |
| ● | issued patents that we own or have exclusively licensed may be revoked, modified, or held invalid or unenforceable, as a result of legal challenges by our competitors; | |
| ● | our competitors might conduct research and development activities in countries where we do not have patent rights and then use the information learned from such activities to develop competitive tests and products for sale in our major commercial markets; | |
| ● | we may not develop additional proprietary technologies that are patentable; | |
| ● | we cannot predict the scope of protection of any patent issuing based on our patent applications, including whether the patent applications that we own or in-license will result in issued patents with claims that are directed to our diagnostic tests and product candidates or uses thereof in the United States or in other foreign countries; | |
| ● | there may be significant pressure on the U.S. government and international governmental bodies to limit the scope of patent protection both inside and outside the United States for disease treatments that prove successful, as a matter of public policy regarding worldwide health concerns; | |
| ● | countries other than the United States may have patent laws less favorable to patentees than those upheld by U.S. courts, allowing foreign competitors a better opportunity to create, develop and market competing diagnostic tests and product candidates; | |
| ● | the claims of any patent issuing based on our patent applications may not provide protection against competitors or any competitive advantages, or may be challenged by third parties; and | |
| ● | if enforced, a court may not hold that our patents are valid, enforceable, and infringed. |
| 30 |
Changes in patent law in the United States and other jurisdictions could diminish the value of patents in general, thereby impairing our ability to protect our diagnostic tests and therapeutic product candidates.
As is the case with other biopharmaceutical companies, our success is heavily dependent on intellectual property, particularly patents. Obtaining and enforcing patents in the biopharmaceutical industry involves both technological and legal complexity and is therefore costly, time-consuming and inherently uncertain. Changes in either the patent laws or interpretation of the patent laws in the United States could increase the uncertainties and costs, and may diminish our ability to protect our inventions, obtain, maintain, and enforce our intellectual property rights and, more generally, could affect the value of our intellectual property or narrow the scope of our owned and licensed patents. Patent reform legislation in the United States and other countries, including the Leahy-Smith America Invents Act (the “Leahy-Smith Act”), signed into law on September 16, 2011, could increase those uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents. The Leahy-Smith Act includes a number of significant changes to U.S. patent law. These include provisions that affect the way patent applications are prosecuted, redefine prior art and provide more efficient and cost-effective avenues for competitors to challenge the validity of patents. These include allowing third-party submission of prior art to the USPTO during patent prosecution and additional procedures to attack the validity of a patent by USPTO-administered post-grant proceedings, including post-grant review, inter partes review, and derivation proceedings. Further, because of a lower evidentiary standard in these USPTO post-grant proceedings compared to the evidentiary standard in U.S. federal courts necessary to invalidate a patent claim, a third party could potentially provide evidence in a USPTO proceeding sufficient for the USPTO to hold a claim invalid even though the same evidence would be insufficient to invalidate the claim if first presented in a district court action. Accordingly, a third party may attempt to use the USPTO procedures to invalidate our patent claims that would not have been invalidated if first challenged by the third party as a defendant in a district court action. Thus, the Leahy-Smith Act and its implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents, all of which could have a material adverse effect on our business, financial condition, results of operations and prospects.
After March 2013, under the Leahy-Smith Act, the U.S. transitioned to a first inventor to file system in which, assuming that the other statutory requirements are met, the first inventor to file a patent application will be entitled to the patent on an invention regardless of whether a third party was the first to invent the claimed invention. A third party that files a patent application in the USPTO after March 2013, but before we file an application covering the same invention, could therefore be awarded a patent covering an invention of ours even if we had made the invention before it was made by such third party. This will require us to be cognizant going forward of the time from invention to filing of a patent application, but circumstances could prevent us from promptly filing patent applications on our inventions. Since patent applications in the U.S. and most other countries are confidential for a period of time after filing or until issuance, we cannot be certain that we or our licensors were the first to either (i) file any patent application related to our diagnostic tests and therapeutic product candidates and other proprietary technologies we may develop or (ii) invent any of the inventions claimed in our or our licensor’s patents or patent applications. Even where we have a valid and enforceable patent, we may not be able to exclude others from practicing the claimed invention where the other party can show that they used the invention in commerce before our filing date. Thus the Leahy-Smith Act and its implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents, all of which could have a material adverse effect on our business, financial condition, results of operations and prospects.
In addition, the patent positions of companies in the development and commercialization of biologics and pharmaceuticals are particularly uncertain. The U.S. Supreme Court has ruled on several patent cases in recent years, either narrowing the scope of patent protection available in certain circumstances or weakening the rights of patent owners in certain situations. Depending on future actions by the U.S. Congress, the U.S. courts, the USPTO and the relevant law-making bodies in other countries, the laws and regulations governing patents could change in unpredictable ways that would weaken our ability to obtain new patents or to enforce our existing patents and patents that we might obtain in the future. For example, in the 2013 case Assoc. for Molecular Pathology v. Myriad Genetics, Inc., the U.S. Supreme Court held that certain claims to DNA molecules are not patentable. While we do not believe that any of the patents owned or licensed by us will be found invalid based on this decision, we cannot predict how future decisions by the courts, the U.S. Congress or the USPTO may impact the value of our patents.
Obtaining and maintaining patent protection depends on compliance with various procedural, document submissions, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance fees, renewal fees, annuities fees and various other governmental fees on patents and/or patent applications are due to be paid to the USPTO and foreign patent agencies in several stages over the lifetime of the patent and/or patent application. The USPTO and various foreign governmental patent agencies also require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. While an inadvertent lapse can in many cases be cured by payment of a late fee or by other means in accordance with the applicable rules, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. Non-compliance events that could result in abandonment or lapse of a patent or patent application include, but are not limited to, failure to respond to official actions within prescribed time limits, non-payment of fees and failure to properly legalize and submit formal documents. If we fail to maintain the patents and patent applications covering our diagnostic tests or therapeutic product candidates, our competitive position would be adversely affected.
Patent terms may be inadequate to protect our competitive position on our diagnostic tests or therapeutic product candidates for an adequate amount of time.
The term of any individual patent depends on applicable law in the country where the patent is granted. In the United States, provided all maintenance fees are timely paid, a patent generally has a term of 20 years from its application filing date or earliest claimed non-provisional filing date. Extensions may be available under certain circumstances, but the life of a patent and, correspondingly, the protection it affords is limited. Even if we or our licensors obtain patents covering our diagnostic tests and therapeutic product candidates, when the terms of all patents covering a diagnostic test or therapeutic product expire, our business may become subject to competition from competitive medications, including generic medications. Given the amount of time required for the development, testing and regulatory review and approval of new diagnostic test or therapeutic product candidates, patents protecting such candidates may expire before or shortly after such candidates are commercialized. As a result, our owned and licensed patent portfolio may not provide us with sufficient rights to exclude others from commercializing diagnostic tests and therapeutic products similar or identical to ours.
| 31 |
Issued patents covering our product candidates could be found invalid or unenforceable if challenged in court or the USPTO.
If we or a licensee initiate legal proceedings against a third party to enforce a patent covering one of our diagnostic tests or therapeutic product candidates, the defendant could counterclaim that the patent covering our diagnostic tests or therapeutic product candidate, as applicable, is invalid and/or unenforceable. In patent litigation in the United States, defendant counterclaims alleging invalidity and/or unenforceability are commonplace, and there are numerous grounds upon which a third party can assert invalidity or unenforceability of a patent. Third parties may also raise similar claims before administrative bodies in the United States or abroad, even outside the context of litigation. Such mechanisms include re-examination, inter partes review, post grant review, and equivalent proceedings in foreign jurisdictions (e.g., opposition proceedings). Such proceedings could result in revocation or amendment to our patents in such a way that they no longer cover our diagnostic tests or therapeutic product candidates. The outcome following legal assertions of invalidity and unenforceability is unpredictable. With respect to the validity question, for example, we cannot be certain that there is no invalidating prior art, of which we, our patent counsel and the patent examiner were unaware during prosecution. If a defendant were to prevail on a legal assertion of invalidity and/or unenforceability, we would lose at least part, and perhaps all, of the patent protection on our diagnostic tests or therapeutic product candidates. Such a loss of patent protection could have a material adverse impact on our business.
If we do not obtain patent term extension in the United States under the Hatch-Waxman Act and in foreign countries under similar legislation, thereby potentially extending the term of marketing exclusivity for our diagnostic tests or therapeutic product candidates, our business may be harmed.
In the United States, a patent that covers an FDA-approved drug or biologic may be eligible for a term extension designed to restore the period of the patent term that is lost during the premarket regulatory review process conducted by the FDA. Depending upon the timing, duration and conditions of FDA marketing authorization of our diagnostic tests or therapeutic product candidates, one or more of our U.S. patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984 (the “Hatch-Waxman Act”), which permits a patent term extension of up to five years for a patent covering an approved diagnostic test or therapeutic product as compensation for effective patent term lost during diagnostic test or therapeutic product development and the FDA regulatory review process. A patent term extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of diagnostic test or therapeutic product approval, and only claims covering such approved diagnostic test or drug product, a method for using it or a method for manufacturing it may be extended. In Europe, our diagnostic test or therapeutic product candidates may be eligible for term extensions based on similar legislation. In either jurisdiction, however, we may not receive an extension if we fail to apply within applicable deadlines, fail to apply prior to expiration of relevant patents or otherwise fail to satisfy applicable requirements. Even if we are granted such an extension, the duration of such extension may be less than our request. If we are unable to obtain a patent term extension, or if the term of any such extension is less than our request, the period during which we can enforce our patent rights for that product will be in effect shortened and our competitors may obtain approval to market competing diagnostic tests or products sooner. The resulting reduction of years of revenue from applicable diagnostic tests or products could be substantial.
We enjoy only limited geographical protection with respect to certain patents and we may not be able to protect our intellectual property rights throughout the world.
Filing, prosecuting and defending patents covering our diagnostic tests and therapeutic product candidates in all countries throughout the world would be prohibitively expensive, and even in countries where we have sought protection for our intellectual property, such protection can be less extensive than those in the United States. The requirements for patentability may differ in certain countries, particularly developing countries, and the breadth of patent claims allowed can be inconsistent. In addition, the laws of some foreign countries do not protect intellectual property rights to the same extent as federal and state laws in the United States. In-licensing patents covering our diagnostic tests and therapeutic product candidates in all countries throughout the world may similarly be prohibitively expensive, if such opportunities are available at all. And in-licensing or filing, prosecuting, and defending patents even in only those jurisdictions in which we develop or commercialize our diagnostic tests and therapeutic product candidates may be prohibitively expensive or impractical. Competitors may use our and our licensors’ technologies in jurisdictions where we have not obtained patent protection or licensed patents to develop their own diagnostic tests and therapeutic products and further may export otherwise infringing products to territories where we and our licensors have patent protection, but where enforcement is not as strong as that in the United States or Europe. These diagnostic tests and products may compete with our diagnostic tests and therapeutic product candidates, and our or our licensors’ patents or other intellectual property rights may not be effective or sufficient to prevent them from competing.
The laws of some jurisdictions do not protect intellectual property rights to the same extent as the laws or regulations in the United States and Europe, and many companies have encountered significant difficulties in protecting and defending proprietary rights in such jurisdictions. Moreover, the legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents, trade secrets or other forms of intellectual property, particularly those relating to biotechnology tests and products, which could make it difficult for us to prevent competitors in some jurisdictions from marketing competing tests and products in violation of our proprietary rights generally. Proceedings to enforce our patent rights in foreign jurisdictions, whether or not successful, are likely to result in substantial costs and divert our efforts and attention from other aspects of our business, and additionally could put at risk our or our licensors’ patents of being invalidated or interpreted narrowly, could increase the risk of our or our licensors’ patent applications not issuing, or could provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate, while damages or other remedies may be awarded to the adverse party, which may be commercially significant. If we prevail, damages or other remedies awarded to us, if any, may not be commercially meaningful. Accordingly, our efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license. Furthermore, while we intend to protect our intellectual property rights in our expected significant markets, we cannot ensure that we will be able to initiate or maintain similar efforts in all jurisdictions in which we may wish to market our diagnostic tests and product candidates. Accordingly, our efforts to protect our intellectual property rights in such countries may be inadequate, which may have an adverse effect on our ability to successfully commercialize our diagnostic tests and product candidates in all of our expected significant foreign markets. If we or our licensors encounter difficulties in protecting, or are otherwise precluded from effectively protecting, the intellectual property rights important for our business in such jurisdictions, the value of these rights may be diminished, and we may face additional competition in those jurisdictions.
In some jurisdictions including European countries, compulsory licensing laws compel patent owners to grant licenses to third parties. In addition, some countries limit the enforceability of patents against government agencies or government contractors. In these countries, the patent owner may have limited remedies, which could materially diminish the value of such patent. If we or any of our licensors are forced to grant a license to third parties under patents relevant to our business, or if we or our licensors are prevented from enforcing patent rights against third parties, our competitive position may be substantially impaired in such jurisdictions.
| 32 |
If our trademarks and trade names are not adequately protected, we may not be able to build name recognition in our markets of interest and our business may be adversely affected.
Our current or future trademarks or trade names may be challenged, infringed, circumvented, declared generic or descriptive or determined to be infringing on other marks. We may not be able to protect our rights to these trademarks and trade names or may be forced to stop using these names, which we need for name recognition by potential partners or customers in our markets of interest. During trademark registration proceedings, we may receive rejections of our applications by the USPTO or in other foreign jurisdictions.
Although we would be given an opportunity to respond to those rejections, we may be unable to overcome such rejections. In addition, in the USPTO and in comparable agencies in many foreign jurisdictions, third parties are given an opportunity to oppose pending trademark applications and to seek to cancel registered trademarks. Opposition or cancellation proceedings may be filed against our trademarks, and our trademarks may not survive such proceedings. If we are unable to establish name recognition based on our trademarks and trade names, we may not be able to compete effectively and our business may be adversely affected. We may license our trademarks and tradenames to third parties, such as distributors. Although these license agreements may provide guidelines for how our trademarks and tradenames may be used, a breach of these agreements or misuse of our trademarks and tradenames by our licensees may jeopardize our rights in or diminish the goodwill associated with our trademarks and trade names.
Moreover, any name we have proposed to use with our therapeutic product candidate in the United States must be approved by the FDA, regardless of whether we have registered it, or applied to register it, as a trademark. The FDA typically conducts a review of proposed product names, including an evaluation of potential for confusion with other product names. If the FDA (or an equivalent administrative body in a foreign jurisdiction) objects to any of our proposed proprietary product names, we may be required to expend significant additional resources in an effort to identify a suitable substitute name that would qualify under applicable trademark laws, not infringe the existing rights of third parties and be acceptable to the FDA. Furthermore, in many countries, owning and maintaining a trademark registration may not provide an adequate defense against a subsequent infringement claim asserted by the owner of a senior trademark. At times, competitors or other third parties may adopt trade names or trademarks similar to ours, thereby impeding our ability to build brand identity and possibly leading to market confusion. In addition, there could be potential trade name or trademark infringement claims brought by owners of other registered trademarks or trademarks that incorporate variations of our registered or unregistered trademarks or trade names. If we assert trademark infringement claims, a court may determine that the marks we have asserted are invalid or unenforceable, or that the party against whom we have asserted trademark infringement has superior rights to the marks in question. In this case, we could ultimately be forced to cease use of such trademarks.
Risks Related to Government Regulations
CyPath® Lung is currently being offered as an LDT by PPLS. Should the FDA disagree that CyPath® Lung is an LDT, or if the FDA’s regulatory approach to LDTs should change in the future, our commercialization strategy may be adversely affected, which would negatively affect our results of operations and financial condition.
The FDA considers an LDT to be a test that is developed, validated, and performed within a single laboratory. The FDA has historically asserted its authority to regulate LDTs as medical devices under the Federal Food, Drug, and Cosmetic Act (the “FDCA”), but it has generally exercised enforcement discretion with regard to LDTs. This means that even though the FDA believes it can impose regulatory requirements on LDTs, such as requirements to obtain premarket approval, de novo classification, or clearance of LDTs, it has generally chosen not to enforce those requirements. The FDA has, on occasion, sent warning letters to laboratories offering LDTs that the agency believed were not eligible for enforcement discretion because of how they were developed, validated, performed or marketed and consequent risks to the public.
FDA has indicated that it may seek to increase its regulation of LDTs through rulemaking. The Spring 2023 Agenda of Regulatory and Deregulatory Actions, published semi-annually by the Office of Management and Budget (OMB), indicates that FDA plans to issue a Notice of Proposed Rulemaking in the Federal Register in August 2023 “to make explicit that laboratory developed tests (LDTs) are devices under the Federal Food, Drug, and Cosmetic Act,” although it is unclear whether this will occur. Any future rulemaking, guidance, or other oversight of LDTs and clinical laboratories that develop and perform them, if and when finalized, may affect the sales of our products and how customers use our products, and may require us to change our business model in order to maintain compliance with these laws.
There have been numerous legislative proposals to clarify the FDA’s regulatory authority over medical devices. In 2021, two bills were reintroduced: the Verifying Accurate, Leading-edge IVCT Development Act of 2020 (the “VALID Act”), which would have expressly granted the FDA authority to regulate LDTs under a risk-based framework; and the Verified Innovative Testing in American Laboratories Act of 2020 (the “VITAL Act”), which would have assigned LDTs to regulation solely under CLIA and would have directed CMS to update its CLIA regulations. Neither of these bills were enacted. The VALID Act was reintroduced in March 2023. We cannot predict if either of these bills will be enacted in their current (or any other) form and cannot quantify the effect of these bills on our business. In the meantime, the regulation by the FDA of LDTs remains uncertain.
Enactment of legislation directing FDA to regulate LDTs or promulgation of new regulations for LDT oversight by FDA could materially and adversely affect our business, financial condition and results of operations. If FDA premarket review, classification or approval is required for CyPath® Lung before we obtain de novo classification, our phased strategy for market entry would be adversely affected. Our laboratory licensee, PPLS, could be forced to stop offering CyPath® Lung as an LDT while we work to obtain de novo classification. Our business, results of operations and financial condition would be negatively affected unless and until such review were completed and our request for de novo classification were granted.
Although we do intend to conduct clinical trials in order to receive de novo classification from the FDA as a Class II in vitro diagnostic, there can be no assurance that the trial will have favorable results or that it will generate the results necessary to obtain such clearance.
Delay by or failure of the FDA to grant our request for de novo classification, or failure on our part to comply with applicable requirements, would adversely affect our business, results of operations and financial condition.
The FDCA requires that medical devices introduced to the United States market, unless exempted by regulation, be authorized by the FDA pursuant to either the premarket notification pathway, known as 510(k) clearance, the de novo classification pathway, or the Premarket Approval (“PMA”) pathway. We plan to seek de novo classification for the CyPath® Lung test in the second quarter of 2026. The FDA may not agree that agree that CyPath® Lung meets the criteria for de novo classification, in which case we would be required to submit a PMA to obtain marketing authorization, which would require manufacturing information and a pre-approval inspection of the manufacturing facilities and could require review by an FDA advisory panel comprised of experts outside the FDA. Any delay by or failure of the FDA to grant our de novo request or PMA could adversely affect our consolidated revenues, results of operations and financial condition.
Additionally, obtaining FDA marketing authorization, approval or de novo classification for diagnostics can be expensive, time-consuming and uncertain, and for higher-risk devices can take several years and requires detailed and comprehensive scientific and clinical data. In addition, medical devices are subject to ongoing FDA obligations and continued regulatory oversight and review. Ongoing compliance with FDA regulations increases the cost of conducting our business and subjects us to heightened regulation by the FDA and penalties for failure to comply with these requirements.
| 33 |
Failure by us or our laboratory licensee to comply with applicable laws pertaining to LDTs or IVDs could adversely affect our business, results of operations and financial condition.
The clinical laboratory testing sector is highly regulated in the United States. Our laboratory licensee, PPLS, is accredited by CAP and holds a CLIA certificate of accreditation. Any failure by our laboratory licensee to comply with CLIA/CAP requirements could result in adverse findings on inspection that, if not timely corrected, could result in loss of accreditation and the inability to perform laboratory testing.
Additionally, certain states, including California, Maryland, Nevada, Pennsylvania, and Rhode Island, require laboratories testing specimens from their jurisdictions to hold an out-of-state laboratory license or permit. New York is exempt from, and imposes requirements in addition to, CLIA, including a requirement for test-specific permits of LDTs before they can be used to test specimens from patients in New York. The failure of our laboratory licensee to obtain state licenses or permits, where required, could interfere with our strategy for a national rollout of CyPath® Lung.
ICU Medical is providing the acapella® Choice Blue device to assist patients in expelling sputum out of the lungs into a collection cup noninvasively. This device is 510(k) cleared as a positive expiratory pressure device to help mobilize lung secretions in people with certain lung conditions. The device does not have a cleared indication for use as a specimen collection device. Promotion of the device by us or our partners for use of the device for specimen collection could cause the FDA to consider the device to be adulterated or misbranded in violation of the FDCA, and to require a 510(k) clearance for a specimen collection indication as a condition of distributing the device. Any disruption to our ability to distribute the acapella® Choice Blue could interfere with our ability to collect adequate patient samples necessary for CyPath® Lung.
CyPath® Lung also relies on a proprietary algorithm, which is currently licensed to PPLS and used by PPLS to develop and validate software integrated into the test procedure that generates the quantitative and qualitative diagnostic results that are included in their laboratory report. Certain types of standalone diagnostics software are subject to FDA regulation as a medical device (specifically, software as a medical device or “SaMD”). Some types of SaMD are subject to premarket authorization requirements. If the FDA were to conclude that we or our laboratory licensee is required to obtain premarket authorization for the software, our ability to offer CyPath® Lung as an LDT could be delayed or prevented, which would adversely affect our business.
The third-party licensors of our future therapeutic products, when ready, may be unable to obtain regulatory approval. The denial or delay of any such approval would delay commercialization of our future therapeutic products and have a material adverse effect on our potential to generate revenue, our business and our results of operations.
We plan to license our therapeutic candidates to third parties for development including clinical testing, manufacturing, labeling, packaging, approval, promotion, advertising, storage, recordkeeping, marketing, distribution, post-approval monitoring and reporting, and export and import. These activities that are to be undertaken by third-party licensees of our future therapeutic products are subject to extensive regulation by the FDA, and by foreign health authorities in other countries. These regulations differ from country to country. In the United States, we are not permitted to market our therapeutic product candidates until we receive regulatory approval from the FDA. The process of obtaining regulatory approval is expensive, often takes many years following research and development, and thereafter the commencement of clinical trials and can vary substantially based upon the type, complexity and novelty of the product candidates involved, as well as the target indications and patient population. Despite the time and expense invested in clinical development of product candidates, regulatory approval is never guaranteed. For our licensors to gain approval to market our product candidates, they must provide clinical data that adequately demonstrate the safety and efficacy of the product for the intended indication. We or any third party has not yet obtained regulatory approval to market any of our product candidates in the United States or any other country. Our business depends upon licensing our therapeutic products to third-party pharmaceutical companies that would obtain these regulatory approvals. The FDA can delay, limit or deny approval of these product candidates for many reasons, including:
| ● | the inability of our licensors to satisfactorily demonstrate that the product candidates have acceptable safety and efficacy profiles for the requested indication; | |
| ● | the FDA’s disagreement with the trial designs of our licensors or the interpretation of data from preclinical studies or clinical trials; | |
| ● | the population studied in the clinical trial may not be sufficiently broad or representative to assess safety in the full population for which we seek approval; | |
| ● | the licensors’ inability to demonstrate that clinical or other benefits of our product candidates outweigh any safety or other perceived risks; | |
| ● | the FDA’s determination that additional preclinical or clinical trials are required; | |
| ● | the FDA’s non-approval of the formulation, labeling or specifications of our product candidates; | |
| ● | the FDA’s failure to accept the manufacturing processes, drug product characteristics or facilities of third-party manufacturers with which we or the third-party licensors contract; or | |
| ● | the potential for approval policies or regulations of the FDA to significantly change in a manner rendering clinical data related to any therapeutic product candidate insufficient for approval. |
Even if clinical testing approval of any regulatory filing for our product candidates eventually is completed, the FDA may grant approval contingent on the performance of costly additional post-approval clinical trials. The FDA may also approve our product candidates for a more limited indication or a narrower patient population than the third party originally requested, and the FDA may not approve the labeling that we believe is necessary or desirable for the successful commercialization of our product candidates. If the FDA requires the licensors to narrow the indications to smaller patient subsets, the market opportunities for our product candidates, if approved, and the ability to generate revenues and royalties may be materially limited. To the extent the licensors seeks regulatory approval in foreign countries, they may face challenges similar to those described above with regulatory authorities in applicable jurisdictions.
| 34 |
Obtaining and maintaining regulatory approval of our diagnostic tests or therapeutic product candidates in one jurisdiction does not mean that we will be successful in obtaining regulatory approval of our product candidates in other jurisdictions. Failure to obtain regulatory approval in foreign jurisdictions would prevent our product candidates from being marketed abroad.
In addition to regulations in the United States, to market and sell our diagnostic tests and therapeutic products in the EU, many Asian countries and other jurisdictions, we must obtain separate regulatory approvals and comply with numerous and varying regulatory requirements, both from a clinical and manufacturing perspective. Approval by the FDA does not ensure approval by regulatory or payor authorities in other countries or jurisdictions, and approval by one regulatory or payor authority outside the United States does not ensure approval by regulatory authorities in other countries or jurisdictions or by the FDA. However, a failure or delay in obtaining regulatory approval in one jurisdiction may have a negative effect on the regulatory approval process in others. For example, even if the FDA grants marketing authorization of a diagnostic test or therapeutic product candidate, comparable regulatory authorities in foreign jurisdictions must also approve the manufacturing, marketing and promotion of the diagnostic test or therapeutic product candidate in those countries. Approval procedures vary among jurisdictions and can involve requirements and administrative review periods different from, and greater than, those in the United States, including additional preclinical studies or clinical trials as clinical trials conducted in one jurisdiction may not be accepted by regulatory authorities in other jurisdictions. In many jurisdictions outside the United States, a diagnostic test or therapeutic product candidate must be approved for reimbursement before it can be approved for sale in that jurisdiction. In some cases, the price that we intend to charge for our diagnostic tests or therapeutic products is also subject to approval. A diagnostic test or therapeutic product candidate that has been approved for sale in a particular country may not receive reimbursement approval in that country. We may not be able to obtain approvals from regulatory authorities or payor authorities outside the United States on a timely basis, if at all.
We may also submit marketing applications in other countries, such as countries in Europe or Asia. We may not be able to file for regulatory approvals and may not receive necessary approvals to commercialize our diagnostic tests or therapeutic products in any jurisdiction. Regulatory authorities in jurisdictions outside of the United States have requirements for approval of diagnostic tests or therapeutic product candidates with which we must comply prior to marketing in those jurisdictions. Obtaining foreign regulatory approvals and compliance with foreign regulatory requirements could result in significant delays, difficulties and costs for us and could delay or prevent the introduction of our diagnostic tests or therapeutic products in certain countries. We do not have any diagnostic tests or therapeutic product candidates approved for sale in any foreign jurisdiction, including international markets, and we do not have experience in obtaining regulatory approval in international markets. If we are unable to obtain approval of any of our diagnostic tests or therapeutic product candidates by regulatory or payor authorities in the EU, Asia or elsewhere, or if we fail to comply with the regulatory requirements in foreign jurisdictions, the commercial prospects of that diagnostic test or therapeutic product candidate may be significantly diminished, and our target market will be reduced and our ability to realize the full market potential of our diagnostic tests or therapeutic product candidates will be harmed.
Even if we obtain FDA approval of any of our diagnostic tests or therapeutic product candidates, we may never obtain approval or commercialize such products outside of the United States, which would limit our ability to realize their full market potential.
In order to market any diagnostic test or therapeutic product outside of the United States, we must establish and comply with numerous and varying regulatory requirements of other countries regarding safety and efficacy. Clinical trials conducted in one country may not be accepted by regulatory authorities in other countries, and regulatory approval in one country does not mean that regulatory approval will be obtained in any other country. Approval procedures vary among countries and can involve additional diagnostic and therapeutic product testing and validation and additional administrative review periods. Seeking foreign regulatory approvals could result in significant delays, difficulties and costs for us and may require additional preclinical studies or clinical trials, which would be costly and time-consuming. Regulatory requirements can vary widely from country to country and could delay or prevent the introduction of our diagnostic tests or therapeutic products in those countries. Satisfying these and other regulatory requirements is costly, time-consuming, uncertain and subject to unanticipated delays. In addition, our failure to obtain regulatory approval in any country may delay or have negative effects on the process for regulatory approval in other countries. We do not have any diagnostic test or therapeutic product candidate approved for sale in any jurisdiction, including international markets, and we do not have experience in obtaining regulatory approval in international markets. If we fail to comply with regulatory requirements in international markets or fail to obtain and maintain required approvals, our ability to realize the full market potential of our diagnostic tests or therapeutic products will be harmed.
The impact of recent healthcare reform legislation and other changes in the healthcare industry and in healthcare spending on us is currently unknown, and may adversely affect our business model.
Our revenue prospects could be affected by changes in healthcare spending and policy in the United States and abroad. We operate in a highly regulated industry and new laws, regulations or judicial decisions, or new interpretations of existing laws, regulations or decisions, related to healthcare availability, the method of delivery or payment for healthcare tests, products and services could negatively impact our business, operations and financial condition.
There have been, and likely will continue to be, legislative and regulatory proposals at the foreign, federal and state levels directed at broadening the availability of healthcare and containing or lowering the cost of healthcare, including proposals aimed at lowering prescription drug prices and increasing competition for prescription drugs, as well as additional regulation on pharmaceutical transparency and reporting requirements, any of which could negatively impact our future profitability and increase our compliance burden. We cannot predict the initiatives that may be adopted in the future, including future challenges or significant revisions to the Affordable Care Act. The continuing efforts of the government, insurance companies, managed care organizations and other payors of healthcare services to contain or reduce costs of healthcare and/or impose price controls may adversely affect:
| ● | the demand for our diagnostic tests or therapeutic product candidates, if we or our licensors obtain regulatory approval; | |
| ● | the ability to set a price that we believe is fair for our diagnostic tests and therapeutic products; | |
| ● | the ability to obtain coverage and reimbursement approval for a diagnostic test and therapeutic product; | |
| ● | our ability to generate revenue and achieve or maintain profitability; | |
| ● | the level of taxes that we are required to pay; and | |
| ● | the availability of capital. |
Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payors, which may adversely affect our future profitability.
| 35 |
Risks Related to Ownership of Our Common Stock and Warrants
We do not expect to pay dividends in the foreseeable future. Any return on investment may be limited to the value of our Common Stock.
We do not anticipate paying cash dividends on our Common Stock in the foreseeable future. The payment of dividends on our Common Stock will depend on earnings, financial condition, and other business and economic factors affecting it at such time as our Board of Directors (our “Board”) may consider relevant. If we do not pay dividends, our Common Stock may be less valuable because a return on your investment will occur only if our stock price appreciates.
The Warrants may not have any value.
Each Warrant will have an exercise price equal to $[●]. The Warrants will be exercisable, in whole or in part, commencing on the date that is 180 days from the commencement of the sales of the Units and will expire on the fifth anniversary of the effective date of the registration statement related to the Offering. In the event our Common Stock price does not exceed the exercise price of the Warrants during the period when the Warrants are exercisable, the Warrants may not have any value.
Holders of Warrants have no rights as stockholders until such holders exercise their Warrants and acquire our shares of Common Stock.
Until holders of our Warrants acquire shares of Common Stock upon exercise thereof, such holders will have no rights with respect to the shares of Common Stock underlying the Warrants. Upon exercise of the Warrants, the holders will be entitled to exercise the rights of a stockholder only as to matters for which the record date occurs after the date they were entered in the register of members of the Company as a stockholder.
The Warrant certificates governing our Warrants designate the state and federal courts of the State of New York sitting in the City of New York, Borough of Manhattan, as the exclusive forum for actions and proceedings with respect to all matters arising out of the Warrants, which could limit a Warrant holder’s ability to choose the judicial forum for disputes arising out of the Warrants.
The warrant certificates governing our Warrants provide that all legal proceedings concerning the interpretations, enforcement and defense of the transactions contemplated by the warrant certificate (whether brought against a party to the warrant certificate or their respective affiliates, directors, officers, shareholders, partners, members, employees or agents) shall be commenced exclusively in the state and federal courts sitting in the City of New York. The warrant certificates further provide that we and the Warrant holders irrevocably submit to the exclusive jurisdiction of the state and federal courts sitting in the City of New York, Borough of Manhattan for the adjudication of any dispute under the warrant certificate or in connection with it or with any transaction contemplated by it or discussed in it. Furthermore, we and the Warrant holders irrevocably waive, and agree not to assert in any suit, action or proceeding, any claim that we or they are not personally subject to the jurisdiction of any such court, that such suit, action or proceeding is improper or is an inconvenient venue for such proceeding. With respect to any complaint asserting a cause of action arising under the Securities Act or the rules and regulations promulgated thereunder, we note, however, that there is uncertainty as to whether a court would enforce this provision and that investors cannot waive compliance with the federal securities laws and the rules and regulations thereunder. Section 22 of the Securities Act creates concurrent jurisdiction for state and federal courts over all suits brought to enforce any duty or liability created by the Securities Act or the rules and regulations thereunder. Section 27 of the Exchange Act creates exclusive federal jurisdiction over all suits brought to enforce any duty or liability created by the Exchange Act or the rules and regulations thereunder. As a result, the exclusive forum provision in the warrant certificates expressly does not apply to suits brought to enforce any duty or liability created by the Exchange Act.
Any person or entity purchasing or otherwise acquiring or holding or owning (or continuing to hold or own) any interest in any of our Warrants shall be deemed to have notice of and consented to the foregoing provisions. Although we believe this exclusive forum provision benefits us by providing increased consistency in the application of the governing law in the types of lawsuits to which it applies, the exclusive forum provision may limit a Warrant holder’s ability to bring a claim in a judicial forum of its choosing for disputes with us or any of our directors, officers, other employees, stockholders, or others which may discourage lawsuits with respect to such claims. Our Warrant holders will not be deemed to have waived our compliance with the federal securities laws and the rules and regulations thereunder as a result of this exclusive forum provision. Further, in the event a court finds the exclusive forum provision contained in our Warrant certificates to be unenforceable or inapplicable in an action, we may incur additional costs associated with resolving such action in other jurisdictions, which could harm our results of operations.
We will have broad discretion in the use of the net proceeds of this Offering and may not use them effectively or in ways that increase the value of our share price.
While we believe that our use of the net proceeds that we will receive from this Offering will be accomplished, we cannot assure you that circumstances will not result in a change of such use. As a result, we will have discretion in the application of the net proceeds, including working capital and other general corporate purposes, and you and other stockholders may disagree with how we spend or invest these proceeds. The failure by our management to apply these funds effectively could adversely affect our business and financial condition. Pending their use, we may invest the net proceeds from our Offering in a manner that does not produce income or that loses value. These investments may not yield a favorable return to our investors.
Future sales of substantial amounts of shares of our Common Stock by existing shareholders could adversely affect the trading price of our Common Stock and Warrants.
If our existing shareholders sell substantial amounts of shares of our Common Stock following the Offering, the market price of our Common Stock and Warrants could fall. In addition, exercise of currently outstanding warrants or options could impact the market price of our Common Stock. Such sales by our existing shareholders might make it more difficult for us to issue new equity or equity-related securities in the future at a time and place we deem appropriate. The shares of Common Stock and the Warrants offered in this Offering will be eligible for immediate resale in the public market without restrictions. All remaining shares of Common Stock, which are currently held by our existing shareholders, may be sold in the public market in the future subject to the lock-up agreements and the restrictions contained in Rule 144 under the Securities Act of 1933, as amended (the “Securities Act”). If any existing shareholders sell a substantial amount of shares, the prevailing market price for our Common Stock could be adversely affected.
| 36 |
If you invest in securities in this Offering, you will incur immediate and substantial dilution in the book value of your Common Stock.
The Offering Price per share of our Common Stock that is part of a Unit will be substantially higher than the net tangible book value per share of our Common Stock immediately after this Offering. Investors purchasing Units in this Offering will pay a price per Unit that substantially exceeds the book value of our tangible assets after subtracting our liabilities. As a result, investors purchasing Units in this Offering will incur immediate dilution of $0.72 per share of our Common Stock, based on the assumed Offering Price of $1.62 per Unit.
This dilution is due to our investors who purchased shares of our Common Stock prior to this Offering having paid substantially less when they purchased their shares than the price offered to the public in this Offering and the exercise of stock options granted to our employees. To the extent that our convertible notes, bridge notes, or Preferred Stock shares are converted into Common Stock or outstanding warrants or stock options are exercised, we issue restricted stock to our employees under our equity incentive plan, or if we otherwise issue additional shares of our Common Stock in each case at per share prices below the price to the public in this Offering, there will be further dilution to new investors. As a result of the dilution to investors purchasing Units in this Offering, investors may receive significantly less than the purchase price paid in this Offering, if anything, in the event of our liquidation. For a further description of the dilution that you will experience immediately after this Offering, see “Dilution.”
The financial and operational projections that we may make from time to time are subject to inherent risks.
The projections that we provide herein or our management may provide from time to time (including, but not limited to, those relating to potential peak sales amounts, clinical and regulatory timelines, production and supply matters, commercial launch dates, and other financial or operational matters) reflect numerous assumptions made by management, including assumptions with respect to our specific as well as general business, regulatory, economic, market, and financial conditions and other matters, all of which are difficult to predict and many of which are beyond our control. Accordingly, there is a risk that the assumptions made in preparing the projections, or the projections themselves, will prove inaccurate. There may be differences between actual and projected results, and actual results may be materially different from those contained in the projections.
Our failure to meet the continued listing requirements of The Nasdaq Capital Market could result in a de-listing of our common stock.
Our shares of our Common Stock are listed for trading on The Nasdaq Capital Market under the symbol “BIAF and our Tradeable Warrants are listed under the symbol “BIAFW.” If we fail to satisfy the continued listing requirements of The Nasdaq Capital Market, such as the corporate governance requirements, the stockholder’s equity requirement or the minimum closing bid price requirement, The Nasdaq Capital Market may take steps to de-list our common stock or warrants. Such a de-listing or even notification of failure to comply with such requirements would likely have a negative effect on the price of our common stock and warrants and would impair your ability to sell or purchase our common stock when you wish to do so. In the event of a de-listing, we would take actions to restore our compliance with The Nasdaq Capital Market’s listing requirements, but we can provide no assurance that any such action taken by us would allow our common stock become listed again, stabilize the market price or improve the liquidity of our common stock, prevent our common stock from dropping below The Nasdaq Capital Market, minimum bid price requirement or prevent future non-compliance with The Nasdaq Capital Market’s listing requirements.
The National Securities Markets Improvement Act of 1996, which is a federal statute, prevents or preempts the states from regulating the sale of certain securities, which are referred to as “covered securities.” Because our common stock is listed on The Nasdaq Capital Market, our common stock is a covered security. Although the states are preempted from regulating the sale of covered securities, the federal statute does allow the states to investigate companies if there is a suspicion of fraud, and, if there is a finding of fraudulent activity, then the states can regulate or bar the sale of covered securities in a particular case. Further, if we were to be delisted from The Nasdaq Capital Market, our common stock would cease to be recognized as a covered security and we would be subject to regulation in each state in which we offer our securities.
Our stock price has fluctuated in the past, has recently been volatile and may be volatile in the future, and as a result, investors in our common stock could incur substantial losses.
Investors should consider an investment in our common stock risky and invest only if they can withstand a significant loss and wide fluctuations in the market value of their investment. Investors who purchase our common stock may not be able to sell their shares at or above the purchase price. Our stock price has been volatile and may be volatile in the future. In addition, the ongoing COVID-19 pandemic has caused broad stock market and industry fluctuations. The stock market in general has been, and the market price of our Common Stock or Warrants in particular, will likely be subject to fluctuation, whether due to, or irrespective of, our operating results and financial condition. The market price of our Common Stock or Warrants may fluctuate as a result of a number of factors, some of which are beyond our control, including, but not limited to:
| ● | actual or anticipated variations in our and our competitors’ results of operations and financial condition; | |
| ● | market acceptance of our diagnostic tests and therapeutic products; | |
| ● | the mix of products that we sell and related services that we provide; | |
| ● | changes in earnings estimates or recommendations by securities analysts, if our Common Stock is covered by analysts; | |
| ● | development of technological innovations or new competitive diagnostic tests or therapeutic products by others; | |
| ● | announcements of technological innovations or new diagnostic tests or therapeutic products by us; | |
| ● | our failure to achieve a publicly announced milestone; | |
| ● | delays between our expenditures to develop and market new or enhanced diagnostic tests or therapeutic products and the generation of sales from those diagnostic tests and therapeutic products; | |
| ● | developments concerning intellectual property rights, including our involvement in litigation; |
| 37 |
| ● | regulatory developments and the decisions of regulatory authorities as to the approval or rejection of new or modified diagnostic tests or therapeutic products; | |
| ● | changes in the amounts that we spend to develop, acquire, or license new diagnostic tests or therapeutic products, technologies, or businesses; | |
| ● | changes in our expenditures to promote our diagnostic tests or therapeutic products; | |
| ● | our sale or proposed sale, or the sale by our significant shareholders, of our Common Stock or other securities in the future; | |
| ● | changes in key personnel; | |
| ● | success or failure of our research and development projects or those of our competitors; | |
| ● | the trading volume of our Common Stock; and | |
| ● | general economic and market conditions and other factors, including factors unrelated to our operating performance. |
These factors and any corresponding price fluctuations may materially and adversely affect the market price of our Common Stock or Warrants and result in substantial losses being incurred by our investors. In the past, following periods of market volatility, public company shareholders have often instituted securities class action litigation. If we were involved in securities litigation, it could impose a substantial cost upon us and divert the resources and attention of our management from our business.
Our Common Stock has often been thinly traded, so investors may be unable to sell at or near ask prices or at all if investors need to sell shares to raise money or otherwise desire to liquidate their shares.
To date, there have been many days on which limited trading of our common stock took place. We cannot predict the extent to which investors’ interests will lead to an active trading market for our common stock or whether the market price of our common stock will be volatile. If an active trading market does not develop, investors may have difficulty selling our common stock. We are likely to be too small to attract the interest of many brokerage firms and analysts. We cannot give investors any assurance that an active public trading market for our common stock will develop or be sustained. The market price of our common stock could be subject to wide fluctuations in response to quarterly variations in our revenues and operating expenses, announcements of new products or services by us, significant sales of our common stock, including “short” sales, the operating and stock price performance of other companies that investors may deem comparable to us, and news reports relating to trends in our markets or general economic conditions.
An investment in our Company may involve tax implications, and you are encouraged to consult your own advisors as neither we nor any related party is offering any tax assurances or guidance regarding our Company or your investment.
The formation of our Company, as well as an investment in our Company generally, involves complex federal, state, and local income tax considerations. Neither the Internal Revenue Service nor any state or local taxing authority has reviewed the transactions described herein and may take different positions than the ones contemplated by management. You are strongly urged to consult your own tax and other advisors prior to investing, as neither we nor any of our officers, directors, or related parties can offer tax or similar advice, nor are any such persons making any representations and warranties regarding such matters.
Our ability to use our net operating loss carry-forwards and certain other tax attributes may be limited.
Under Section 382 of the Internal Revenue Code of 1986, as amended, if a corporation undergoes an “ownership change” (generally defined as a greater than 50% change (by value) in its equity ownership over a three-year period), the corporation’s ability to use its pre-change net operating loss carry-forwards and other pre-change tax attributes (such as research tax credits) to offset its post-change income may be limited. We may experience ownership changes in the future as a result of subsequent shifts in our stock ownership, including the completion of our offering taken together with other transactions we may consummate in the succeeding three-year period. As a result, if we earn net taxable income, our ability to use our pre-change net operating loss carry-forwards to offset U.S. federal taxable income may be subject to limitations, which potentially could result in increased future tax liability.
Our Certificate of Incorporation permits “blank check” Preferred Stock, which can be designated by our Board without stockholder approval.
We are authorized to issue 20,000,000 shares of Preferred Stock. The shares of our Preferred Stock may be issued from time to time in one or more series, each of which shall have a distinctive designation or title as is determined by our Board prior to the issuance of any shares thereof. The Preferred Stock may have such voting powers, full, enhanced or limited, or no voting powers, and such preferences and relative, participating, optional, or other special rights and such qualifications, limitations, or restrictions thereof as adopted by the Board, which may include enhanced dividend rights, rights of redemption, sinking funds to pay dividends, liquidation and other rights that would be different than, and preferential to, the rights of the Common Stockholders. Because our Board is able to designate the powers and preferences of the Preferred Stock without the vote of a majority of our stockholders, Common Stockholders will have no control over what designations and preferences our Preferred Stock will have. If Preferred Stock is designated and issued, then depending upon the designation and preferences, the holders of the Preferred Stock may exercise voting control. As a result, our stockholders would have no control over the operations of our Company.
| 38 |
Provisions in our corporate charter documents and under Delaware law could make an acquisition of us, which may be beneficial to our stockholders, more difficult and may prevent attempts by our stockholders to replace or remove our current management.
Provisions in our certificate of incorporation, as amended (our “Charter”) and amended and restated bylaws (our “A&R Bylaws”) may discourage, delay or prevent a merger, acquisition or other change in control of us that stockholders may consider favorable, including transactions in which you might otherwise receive a premium for your shares. These provisions also could limit the price that investors might be willing to pay in the future for shares of our Common Stock, thereby depressing the market price of our Common Stock. In addition, because our Board is responsible for appointing the members of our management team, these provisions may frustrate or prevent any attempts by our stockholders to replace or remove our current management by making it more difficult for stockholders to replace members of our Board. Among other things, these provisions:
| ● | allow the authorized number of our directors to be changed only by resolution of our Board; | |
| ● | establish advance notice requirements for stockholder proposals that can be acted on at stockholder meetings and nominations to our Board; | |
| ● | require that stockholder actions must be effected at a duly called stockholder meeting and prohibit actions by our stockholders by written consent; | |
| ● | prohibit our stockholders from calling a special meeting of our stockholders; and | |
| ● | authorize our Board to issue Preferred Stock without stockholder approval, which could be used to institute a stockholder rights plan, or so-called “poison pill,” that would work to dilute the stock ownership of a potential hostile acquirer, effectively preventing acquisitions that have not been approved by our Board. |
Moreover, because we are incorporated in Delaware, we are governed by the provisions of Section 203 of the Delaware General Corporation Law (the “DGCL”), which prohibits a person who owns 15% or more of our outstanding voting stock from merging or combining with us for a period of three years after the date of the transaction in which the person acquired 15% or more of our outstanding voting stock, unless the merger or combination is approved in a prescribed manner. These provisions could discourage potential acquisition proposals and could delay or prevent a change in control transaction. They could also have the effect of discouraging others from making tender offers for our Common Stock, including transactions that may be your best interests. These provisions may also prevent changes in our management or limit the price that investors are willing to pay for our stock.
Certain provisions in our Charter and A&R Bylaws could make a merger, tender offer, or proxy contest difficult, thereby depressing the trading price of our Common Stock.
Our Charter and A&R Bylaws contain provisions that could depress the trading price of our Common Stock by acting to discourage, delay or prevent a change of control of our Company or changes in our management that the stockholders of our Company may deem advantageous. These provisions include the following:
| ● | permit the Board to establish the number of directors and fill any vacancies and newly-created directorships; | |
| ● | authorize the issuance of “blank check” preferred stock that our Board could use to implement a stockholder rights plan; | |
| ● | prohibit stockholders from calling special meetings of stockholders; | |
| ● | prohibit stockholder action by written consent, which requires all stockholder actions to be taken at a meeting of our stockholders; | |
| ● | provide that the Board is expressly authorized to adopt, amend, alter or repeal our bylaws; | |
| ● | restrict the forum for certain litigation against us to Delaware; and | |
| ● | establish advance notice requirements for nominations for election to our Board or for proposing matters that can be acted upon by stockholders at annual stockholder meetings. |
Any provision in our Charter or A&R Bylaws that has the effect of delaying or deterring a change in control could limit the opportunity for our stockholders to receive a premium for their shares of our Common Stock and could also affect the price that some investors are willing to pay for our Common Stock.
Certain provisions of the DGCL may have anti-takeover effects that could delay, defer, or discourage another party from acquiring control of us, prevent changes in our Board or management, and make certain transactions more challenging that stockholders might otherwise believe to be in their best interests.
We are subject to the provisions of Section 203 of the DGCL, which generally prohibits us from engaging in a “business combination,” meaning a merger, asset sale, or other transaction resulting in a stockholder’s financial benefit, with an “interested stockholder” for a three-year period following the time that such stockholder becomes an interested stockholder, unless the business combination is approved in a manner prescribed by Section 203. Section 203 defines an “interested stockholder” as a person who, together with affiliates and associates, owns, or within three years did own, 15% or more of a corporation’s outstanding voting stock. These provisions may have the effect of delaying, deferring, or preventing changes in control of our Company and of averting changes in our Board or management. They are expected to discourage certain types of coercive takeover practices and inadequate takeover bids and, as a consequence, they might also inhibit temporary fluctuations in the market price of our Common Stock that often result from actual or rumored hostile takeover attempts. These provisions could make it more difficult to accomplish transactions that stockholders might otherwise deem to be in their best interests.
Our Charter designates a state or federal court located within the state of Delaware as the exclusive forum for substantially all disputes between us and our stockholders, which could limit our stockholders’ ability to choose the judicial forum for disputes with us or our directors, officers or employees.
Our Charter provides that, unless we consent in writing to the selection of an alternative forum, to the fullest extent permitted by law, the sole and exclusive forum for (1) any derivative action or proceeding brought on our behalf, (2) any action asserting a claim of breach of a fiduciary duty owed by any of our directors, officers, stockholder or employees to us or our stockholders, (3) any action asserting a claim arising pursuant to any provision of the DGCL, our Charter or our A&R Bylaws or as to which the DGCL confers jurisdiction on the Court of Chancery of the State of Delaware, shall be the Court of Chancery of the State of Delaware (or, if the Court of Chancery does not have jurisdiction, the federal district court for the District of Delaware) in all cases subject to the court having jurisdiction over indispensable parties named as defendants. These exclusive-forum provisions do not apply to claims under the Securities Act.
| 39 |
Section 27 of the Exchange Act creates exclusive federal jurisdiction over all suits brought to enforce any duty or liability created by the Exchange Act or the rules and regulations thereunder. As a result, the exclusive forum provision will not apply to suits brought to enforce any duty or liability created by the Exchange Act or any other claim for which the federal courts have exclusive jurisdiction. Section 22 of the Securities Act creates concurrent jurisdiction for federal and state courts over all suits brought to enforce any duty or liability created by the Securities Act or the rules and regulations thereunder. However, our Charter and our A&R Bylaws contain a federal forum provision which provides that unless we consent in writing to the selection of an alternative forum, the federal district courts of the United States of America will be the exclusive forum for the resolution of any complaint asserting a cause of action arising under the Securities Act. We note, however, that there is uncertainty as to whether a court would enforce this provision and that investors cannot waive compliance with the federal securities laws and the rules and regulations thereunder.
Any person or entity purchasing or otherwise acquiring any interest in any of our securities shall be deemed to have notice of and consented to this provision. This exclusive forum provision may limit a stockholder’s ability to bring a claim in a judicial forum of its choosing for disputes with us or our directors, officers, or other employees, which may discourage lawsuits against us and our directors, officers, and other employees. If a court were to find the exclusive forum provision in our Charter to be inapplicable or unenforceable in an action, we may incur additional costs associated with resolving the dispute in other jurisdictions, which could harm our results of operations.
Certain limitation-of-liability and indemnification provisions in our Charter and A&R Bylaws may discourage stockholders from bringing a lawsuit against our directors and officers for breaches of their fiduciary duties, may reduce the likelihood of derivative litigation against our directors and officers, even though an action, if successful, might benefit the Company and other stockholders, and may adversely impact stockholders’ investments to the extent that the Company pays the costs of settlement and damage awards against directors and officers as required by these indemnification provisions.
Our Charter contains provisions that limit the liability of our directors for monetary damages to the fullest extent permitted by the DGCL. Consequently, our directors will not be personally liable to us or our stockholders for monetary damages for any breach of fiduciary duties as directors, except liability for:
| ● | any breach of the director’s duty of loyalty to us or our stockholders; | |
| ● | any act or omission not in good faith or that involves intentional misconduct or a knowing violation of law; | |
| ● | unlawful payments of dividends or unlawful stock repurchases or redemptions as provided in Section 174 of the DGCL; or | |
| ● | any transaction from which the director derived an improper personal benefit. |
Our Charter and our A&R Bylaws require us to indemnify our directors and officers, and allow us to indemnify other employees and agents, to the fullest extent permitted by the DGCL. Subject to certain limitations and limited exceptions, our Charter and A&R Bylaws also require us to advance expenses incurred by our directors and officers for the defense of any action for which indemnification is required or permitted.
While we believe that including the limitation-of-liability and indemnification provisions in our Charter, A&R Bylaws, and indemnification agreements is necessary to attract and retain qualified persons such as directors, officers and key employees, those provisions may discourage stockholders from bringing a lawsuit against our directors and officers for breaches of their fiduciary duties. They may also reduce the likelihood of derivative litigation against our directors and officers, even though an action, if successful, might benefit us and other stockholders. Further, a stockholder’s investment may be adversely affected to the extent that we pay the costs of settlement and damage awards against directors and officers as required by these indemnification provisions.
Our management collectively owns a substantial majority of our Common Stock.
Based on the provisions for determining beneficial ownership in accordance with Rule 13d-3 and Item 403 of Regulation S-K under the Exchange Act, immediately after this Offering, our officers and directors will own or exercise control of approximately 32% of the voting power of our outstanding Common Stock. As a result, investors may be prevented from affecting matters involving our Company, including:
| ● | the composition of our Board and, through it, any determination with respect to our business direction and policies, including the appointment and removal of officers; | |
| ● | any determinations with respect to mergers or other business combinations; | |
| ● | our acquisition or disposition of assets; and | |
| ● | our corporate financing activities. |
Furthermore, this concentration of voting power could have the effect of delaying, deterring, or preventing a change of control or other business combination that might otherwise be beneficial to our stockholders. This significant concentration of share ownership may also adversely affect the trading price for our Common Stock because investors may perceive disadvantages in owning stock in a company that is controlled by a small number of stockholders.
If securities or industry analysts do not publish research or publish inaccurate or unfavorable research about our business, our stock price and trading volume could decline.
The trading market for our Common Stock will depend in part on the research and reports that securities or industry analysts publish about us or our business. Securities and industry analysts do not currently, and may never, publish research on our Company. If no or only very few securities analysts commence coverage of us, or if industry analysts cease coverage of us, the trading price for our Common Stock would be negatively affected. If one or more of the analysts who cover us downgrade our Common Stock or publish inaccurate or unfavorable research about our business, our Common Stock price would likely decline. If one or more of these analysts cease coverage of us or fail to publish reports on us regularly, demand for our Common Stock could decrease, which might cause our Common Stock price and trading volume to decline.
| 40 |
If we fail to establish and maintain an effective system of internal control or disclosure controls and procedures are not effective, we may not be able to report our financial results accurately and timely or to prevent fraud. Any inability to report and file our financial results accurately and timely could harm our reputation and adversely impact the trading price of our Common Stock.
Effective internal controls are necessary for us to provide reliable financial reports and effectively prevent fraud. Section 404 of the Sarbanes-Oxley Act of 2002 (the “Sarbanes-Oxley Act”) requires us to evaluate and report on our internal controls over financial reporting and, depending on our future growth, may require our independent registered public accounting firm to annually attest to our evaluation, as well as issue its own opinion on our internal controls over financial reporting. The process of implementing and maintaining proper internal controls and complying with Section 404 is expensive and time consuming. We cannot be certain that the measures we will undertake will ensure that we will maintain adequate controls over our financial processes and reporting in the future. Furthermore, if we are able to rapidly grow our business, the internal controls that we will need may become more complex, and significantly more resources will be required to ensure our internal controls remain effective. Failure to implement required controls or difficulties encountered in their implementation could harm our operating results or cause us to fail to meet our reporting obligations. If we or our auditors discover a material weakness in our internal controls, the disclosure of that fact, even if the weakness is quickly remedied, could diminish investors’ confidence in our financial statements and harm our stock price. In addition, non-compliance with Section 404 could subject us to a variety of administrative sanctions, including the suspension of trading, ineligibility for future listing on one of the Nasdaq Stock Markets or national securities exchanges, and the inability of registered broker-dealers to make a market in our Common Stock, which may reduce our stock price.
General Risks
We are an “emerging growth company” and a “smaller reporting company,” and the reduced disclosure requirements applicable to emerging growth companies and smaller reporting companies may make our Units less attractive to investors.
We are an “emerging growth company” as defined in the JOBS Act, and we intend to take advantage of some of the exemptions from reporting requirements that are applicable to other public companies that are not emerging growth companies, including:
| ● | being permitted to provide only two years of audited financial statements, in addition to any required unaudited interim financial statements, with correspondingly reduced MD&A disclosure; | |
| ● | not being required to comply with the auditor attestation requirements in the assessment of our internal control over financial reporting; | |
| ● | not being required to comply with any requirement that may be adopted by the Public Company Accounting Oversight Board or a supplement to the auditor’s report providing additional information about the audit and the financial statements; | |
| ● | reduced disclosure obligations regarding executive compensation; and | |
| ● | not being required to hold a non-binding advisory vote on executive compensation or obtain stockholder approval of any golden parachute payments not previously approved. |
In addition, as an “emerging growth company” the JOBS Act allows us to delay adoption of new or revised accounting pronouncements applicable to public companies until such pronouncements are made applicable to private companies, unless we later irrevocably elect not to avail ourselves of this exemption. We have elected to use this extended transition period under the JOBS Act. As a result, our financial statements may not be comparable to the financial statements of issuers who are required to comply with the effective dates for new or revised accounting standards that are applicable to public companies, which may make comparison of our financials to those of other public companies more difficult. We will remain an emerging growth company until the earlier of: (i) the last day of the fiscal year (1) following the fifth anniversary of the completion of this Offering, (2) in which we have total annual gross revenue of at least $1.235 billion, or (3) in which we are deemed to be a large accelerated filer, which means the market value of our Common Stock that is held by non-affiliates exceeds $700 million as of the prior June 30th; and (ii) the date on which we have issued more than $1.0 billion in non-convertible debt during the prior three-year period.
We are also a “smaller reporting company” meaning that the market value of our stock held by non-affiliates plus the proposed aggregate amount of gross proceeds to us as a result of this Offering is less than $700 million and our annual revenue was less than $100 million during the most recently completed fiscal year. Smaller reporting companies may take advantage of certain reduced disclosure obligations, including, among other things, providing only two years of audited financial statements in our Annual Report on Form 10-K, and, similar to emerging growth companies, smaller reporting companies have reduced disclosure obligations regarding executive compensation. To the extent we take advantage of such reduced disclosure obligations, it may also make comparison of our financial statements with other public companies difficult or impossible. We will remain a smaller reporting company until the last day of the fiscal year in which (i) the market value of our common shares held by non-affiliates exceeds $250 million as of the end of that year’s second fiscal quarter, or (ii) our annual revenues exceeded $100 million during such completed fiscal year and the market value of our common shares held by non-affiliates exceeds $700 million as of the end of that year’s second fiscal quarter.
Investors may find our find our Common Stock less attractive to the extent we will rely on these exemptions. If some investors find our Common Stock less attractive as a result, there may be a less active trading market for our Common Stock and our stock price may be more volatile.
We have incurred significant increased costs as a result of operating as a public company, and our management is required to devote substantial time to compliance initiatives.
As a public company, we have incurred and will continue to incur legal, accounting and other expenses that we did not incur as a private company. We are subject to the reporting requirements of the Exchange Act and are required to comply with the applicable requirements of the Sarbanes-Oxley Act of 2002 and the Dodd-Frank Wall Street Reform and Consumer Protection Act. The listing requirements of the Nasdaq Stock Market, and the rules of the SEC require that we satisfy certain corporate governance requirements. Our management and other personnel are required to devote a substantial amount of time to ensure that we comply with all of these requirements. Moreover, the reporting requirements, rules and regulations have increased our legal and financial compliance costs and will make some activities more time-consuming and costly. Any changes we make to comply with these obligations may not be sufficient to allow us to satisfy our obligations as a public company on a timely basis, or at all. These reporting requirements, rules and regulations, coupled with the increase in potential litigation exposure associated with being a public company, could also make it more difficult for us to attract and retain qualified persons to serve on our board of directors or board committees or to serve as executive officers, or to obtain certain types of insurance, including directors’ and officers’ insurance, on acceptable terms.
| 41 |
As a public company, we are required to maintain internal control over financial reporting and to report any material weaknesses in such internal controls. Beginning with the second annual report on Form 10-K that we will be required to file with the SEC, Section 404 requires an annual management assessment of the effectiveness of our internal control over financial reporting. The rules governing the standards that must be met for management to assess our internal control over financial reporting are complex and require significant documentation, testing, and possible remediation.
As of June 30, 2023, our Chief Executive Officer and Chief Financial Officer evaluated the effectiveness of our “disclosure controls and procedures” (as defined in the Exchange Act) Rules 13a-15(e) and 15d-15(e)). Based on that evaluation, management has concluded that due to limited resources and limited number of employees, its internal control over financial reporting was ineffective as of June 30, 2023, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements in accordance with generally accepted accounting principles in the U.S. (“GAAP”). To mitigate the limited resources and employees, we rely heavily on direct management oversight of transactions, along with the use of legal and accounting professionals. As we grow, we expect to increase the number of employees, which we believe will enable us to implement adequate segregation of duties within the internal control framework.
In the future, if we identify any additional material weaknesses in our internal control over financial reporting, if we are unable to comply with the requirements of Section 404 of the Sarbanes-Oxley Act in a timely manner, or if we are unable to assert that our internal control over financial reporting is effective, investors may lose confidence in the accuracy and completeness of our financial reports and the market price of our common stock could decline, and we could also become subject to investigations by the stock exchange on which our common stock is listed, the SEC or other regulatory authorities, which could require additional financial and management resources. In addition, as a public company we will be required to file accurate and timely quarterly and annual reports with the SEC under the Exchange Act. Any failure to report our financial results on an accurate and timely basis could result in sanctions, lawsuits, delisting of our shares from Nasdaq or other adverse consequences that would materially harm our business and reputation.
For so long as we remain an emerging growth company as defined in the JOBS Act, we intend to take advantage of certain exemptions from various reporting requirements that are applicable to public companies that are not emerging growth companies, including, but not limited to, not being required to comply with the auditor attestation requirements of Section 404. We will remain an emerging growth company until the earlier of (1) the last day of the fiscal year (i) following the fifth anniversary of the completion of our initial public offering, (ii) in which we have total annual gross revenue of at least $1.235 billion, or (iii) in which we are deemed to be a large accelerated filer, which means the market value of our common stock that is held by non-affiliates exceeds $700.0 million as of the prior June 30th, and (2) the date on which we have issued more than $1.0 billion in non-convertible debt during the prior three-year period.
Our disclosure controls and procedures may not prevent or detect all errors or acts of fraud.
We designed our disclosure controls and procedures to reasonably assure that information we must disclose in reports we file or submit under the Exchange Act is accumulated and communicated to management, and recorded, processed, summarized and reported within the time periods specified in the rules and forms of the SEC. We believe that any disclosure controls and procedures or internal controls and procedures, no matter how well-conceived and operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met.
These inherent limitations include the realities that judgments in decision-making can be faulty, and breakdowns can occur because of simple error or mistake. For example, our directors or executive officers could inadvertently fail to disclose a new relationship or arrangement causing us to fail to make any related party transaction disclosures. Additionally, controls can be circumvented by the individual acts of some persons, by collusion of two or more people or by an unauthorized override of the controls. Accordingly, because of the inherent limitations in our control system, misstatements due to error or fraud may occur and not be detected.
Future changes in financial accounting standards or practices may cause adverse and unexpected revenue fluctuations and adversely affect our reported results of operations.
Future changes in financial accounting standards may cause adverse, unexpected revenue fluctuations and affect our reported financial position or results of operations. Financial accounting standards in the United States are constantly under review and new pronouncements and varying interpretations of pronouncements have occurred with frequency in the past and are expected to occur again in the future. As a result, we may be required to make changes in our accounting policies. Those changes could affect our financial condition and results of operations or the way in which such financial condition and results of operations are reported. We intend to invest resources to comply with evolving standards, and this investment may result in increased general and administrative expenses and a diversion of management time and attention from business activities to compliance activities. See the “MD&A—Recent Accounting Pronouncements” section of this prospectus.
Claims for indemnification by our directors and officers may reduce our available funds to satisfy successful third-party claims against us and may reduce the amount of money available to us.
Our Charter and A&R Bylaws provide that we will indemnify our directors and officers, in each case, to the fullest extent permitted by Delaware law. Delaware law provides that directors of a corporation will not be personally liable for monetary damages for any breach of fiduciary duties as directors, except liability for:
| ● | any breach of the director’s duty of loyalty to the corporation or its stockholders; | |
| ● | any act or omission not in good faith or that involves intentional misconduct or a knowing violation of law; | |
| ● | unlawful payments of dividends or unlawful stock repurchases or redemptions; or | |
| ● | any transaction from which the director derived an improper personal benefit. |
Such limitation of liability does not apply to liabilities arising under federal securities laws and does not affect the availability of equitable remedies such as injunctive relief or rescission.
Our A&R Bylaws provide that we are required to indemnify our directors and officers to the fullest extent permitted by Delaware law and may indemnify our other employees and agents. Our A&R Bylaws also provide that, on satisfaction of certain conditions, we will advance expenses incurred by a director or officer in advance of the final disposition of any action or proceeding, and permit us to secure insurance on behalf of any officer, director, employee or other agent for any liability arising out of his or her actions in that capacity regardless of whether we would otherwise be permitted to indemnify him or her under the provisions of Delaware law. We believe that these Charter and A&R Bylaws provisions are necessary to attract and retain qualified persons as directors and officers.
While we maintain directors’ and officers’ liability insurance, such insurance may not be adequate to cover all liabilities that we may incur, which may reduce our available funds to satisfy third-party claims and may adversely impact our cash position.
| 42 |
USE OF PROCEEDS
We estimate that the net proceeds to us from the sale of Units in this Offering will be approximately $4.0 million, after deducting the underwriting discounts and commissions and estimated offering expenses payable by us. This assumes an Offering Price of $1.62 per Unit. If the underwriters exercise their option to purchase additional shares of our Common Stock in full, the net proceeds to us will be approximately $4.7 million.
We intend to use the net proceeds from this Offering for working capital and for general corporate purposes, which may include product and test development, general and administrative matters, and capital expenditures. We may also use a portion of the net proceeds for the acquisition of, or investment in, technologies, solutions or businesses that complement our business, although we have no present definitive commitments or agreements to enter into any acquisitions or investments. We expect the proceeds from this Offering together with anticipated sales of our diagnostic LDT tests should be sufficient for us to complete the de novo pivotal clinical trial and, if results are positive, to submit and obtain FDA marketing authorization of CyPath® Lung for sale.
We cannot specify with certainty all of the uses of the net proceeds that we will receive from this Offering. Accordingly, we will have broad discretion in the application of these proceeds and our investors will be relying on the judgment of our management regarding the application of the net proceeds of this Offering.
Each $0.25 increase or decrease in the assumed Offering Price of $1.62 per Unit would increase or decrease the net proceeds to us from this Offering by approximately $700,000, assuming that the number of Units offered by us, as set forth on the cover page of this prospectus, remains the same and after deducting underwriting discounts and commissions and the estimated offering expenses payable by us. We may also increase or decrease the number of Units we are offering. Each 250,000 Unit increase or decrease in the number of Units offered by us would increase or decrease the net proceeds to us from this Offering by approximately $150,000, assuming that the assumed Offering Price of $1.62 per Unit remains the same and after deducting underwriting discounts and commissions and the estimated offering expenses payable by us.
| 43 |
DIVIDEND POLICY
We have never declared or paid any cash dividends on our capital stock. We intend to retain all available funds and future earnings, if any, to fund the development and expansion of our business, and we do not anticipate declaring or paying any cash dividends in the foreseeable future. Any future determination regarding the declaration and payment of dividends, if any, will be at the discretion of our Board and will depend on then-existing conditions, including our financial condition, results of operations, contractual restrictions, capital requirements, business prospects, and other factors our Board may deem relevant.
| 44 |
CAPITALIZATION
The following table sets forth our cash and cash equivalents and capitalization as of June 30, 2023:
| ● | on an actual basis, based on 8,555,365 shares of Common Stock issued and outstanding at June 30, 2023, as reflected in our June 30, 2023 unaudited financial statements, which does not include 125,815 shares of unvested restricted Common Stock as of such date; | |
| ● | on a pro forma basis to give effect to: (i) on July 1, 2023, the issuance of an aggregate of 71,715 restricted shares of Common Stock to our seven directors, as part of our director compensation policy; (ii) the issuance of an aggregate of 8,226 shares of Common Stock to a consultant pursuant to the terms of a consulting agreement between July 1, 2023 and September 1, 2023; (iii) the addition of 16,605 shares of restricted Common Stock that were issued but unvested prior to June 30, 2023 and which have subsequently vested; and (iv) the consummation of the Acquisition including the issuance of 564,972 shares of restricted Common Stock to the Joyce Trust pursuant to the terms of the Asset Purchase Agreement, the payment of $2,500,000 cash consideration to Village Oaks, the assets acquired including cash and the liabilities assumed in Acquisition; and | |
| ● | on a pro forma, as adjusted basis to give effect to the pro forma adjustments and the issuance and sale of 3,086,419 Units in this Offering at an assumed Offering Price of $1.62 per Unit, after deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us. |
The information set forth in the table below is illustrative only and our capitalization following the completion of this Offering will be adjusted based on the actual Offering Price, the number of Units sold in this Offering, and other terms of this Offering determined at pricing. You should read the following table in conjunction with our consolidated financial statements and related notes appearing at the end of this prospectus as well as the MD&A and “Description of Securities” sections of this prospectus.
| As of June 30, 2023 | ||||||||||||
| Actual | Pro Forma |
Pro Forma, As Adjusted | ||||||||||
| Cash and cash equivalents | $ | 8,279,182 | $ | 6,161,830 | $ | 10,151,830 | ||||||
| Stockholders’ equity: | ||||||||||||
| Preferred stock, par value $0.001 per share; 20,000,000 shares authorized; no shares issued and outstanding at June 30, 2023, no shares issued and outstanding pro forma, and no shares issued and outstanding pro forma, as adjusted | ||||||||||||
| Common stock, par value $0.007 per share; 25,000,000 shares authorized; 8,555,365 issued and outstanding at June 30, 2023, 9,216,883 shares issued and outstanding pro forma, and 12,303,302 shares issued and outstanding pro forma, as adjusted | 59,887 | 64,518 | 86,123 | |||||||||
| Additional paid-in capital | 47,978,892 | 49,145,303 | 53,113,698 | |||||||||
| Accumulated deficit | (39,940,431 | ) | (40,111,473 | ) | (40,111,473 | ) | ||||||
| Total stockholders’ equity | $ | 8,098,348 | 9,098,348 | 13,088,348 | ||||||||
| Total capitalization | $ | 8,098,348 | 9,098,348 | 13,088,348 | ||||||||
Each $0.25 increase (decrease) in the assumed Offering Price of $1.62 per Unit, would increase (decrease) each of our pro forma, as adjusted cash and cash equivalents, additional paid-in capital, total stockholders’ equity (deficit) and total capitalization by approximately $700,000, assuming that the number of Units offered by us, as set forth on the cover page of this prospectus, remains the same and after deducting the estimated underwriting discounts and commissions and estimated offering expenses. Similarly, each increase (decrease) of 250,000 Units in the number of Units offered by us would increase (decrease) each of our pro forma, as adjusted cash, cash equivalents, additional paid-in capital, total stockholders’ equity (deficit) and total capitalization by approximately $150,000, assuming the assumed Offering Price of $1.62 per Unit remains the same and after deducting the estimated underwriting discounts and commissions and estimated offering expenses.
| (1) | The number of shares of Common Stock to be outstanding immediately following this Offering as represented in this “Capitalization” section is based on 8,555,365 shares of Common Stock outstanding as of June 30, 2023, which does not include 125,815 shares of unvested restricted Common Stock as of such date, and excludes: |
| ● |
133,414 unvested shares of restricted Common Stock as of September 20, 2023, which are outstanding but subject to forfeiture and therefore not included in our balance sheet; |
|
| ● | 3,086,419 shares of Common Stock issuable upon the exercise of the Warrants underlying the Units sold in this Offering; | |
| ● | 462,962 shares of Common Stock issuable upon the exercise of the Over-Allotment Option; | |
| ● | 462,962 shares of Common Stock issuable upon the exercise of 462,963 Warrants issuable upon the exercise of the Over-Allotment Option; | |
| ● | 61,728 shares of Common Stock issuable upon the exercise of the Representative’s Warrant; | |
| ● | 806,392 shares of Common Stock issuable on the exercise of stock options issued under our 2014 Equity Incentive Plan to certain of our employees, directors, and consultants with a weighted average exercise price equal to $4.33; | |
| ● | an aggregate of 4,305,812 shares of Common Stock issuable upon the exercise of outstanding Tradeable Warrants and Non-Tradeable Warrants (which share number reflects an adjustment in the number of shares to be issued upon exercise of the Tradeable Warrants and Non-Tradeable Warrants that will be effected upon consummation of this Offering in accordance with the price protection provisions contained in the Tradeable Warrants and Non-Tradeable Warrants) all of which, upon consummation of the Acquisition, will have an exercise price that will be reduced to $3.0625 per share; | |
| ● | 2,900,904 shares of Common Stock issuable on the exercise of outstanding warrants issued prior to consummation of our initial public offering with a weighted average exercise price equal to $5.31 per share; and. | |
| ● | 658,294 shares of our Common Stock that are reserved for equity awards that may be granted under our 2014 Equity Incentive Plan. |
| 45 |
DILUTION
If you invest in our securities in this Offering, your ownership interest will be immediately diluted to the extent of the difference between the Offering Price per share of our Common Stock that is a part of the Unit and the pro forma as adjusted net tangible book value per share of our Common Stock immediately after this Offering. Net tangible book value dilution per share to new investors represents the difference between the amount per share paid by purchasers of Units in this Offering and the pro forma as adjusted net tangible book value per share of Common Stock immediately after completion of this Offering.
Our historical net tangible book value (deficit) as of June 30, 2023, based on 8,555,365 shares of Common Stock issued and outstanding at June 30, 2023, which does not include 125,815 shares of unvested restricted Common Stock as of such date, was approximately $8.1 million, or $0.95 per share of our Common Stock. Our historical net tangible book value (deficit) is the amount of our total tangible assets, adjusted to remove capitalized deferred offering costs we expect to recognize as an offset to the proceeds from this Offering, less our total liabilities, which is not included within our stockholders’ (deficit) equity. Historical net tangible book value per share represents historical net tangible book value (deficit) divided by 8,555,365, the number of shares of our Common Stock outstanding as of June 30, 2023.
Our pro forma net tangible book value as of June 30, 2023 was $7.1 million, or $0.77 per share of our Common Stock. Pro forma net tangible book value represents the amount of our total tangible assets less our total liabilities, after giving effect to: (i) on July 1, 2023, the issuance of an aggregate of 71,715 restricted shares of Common Stock to our seven directors, as part of our director compensation policy; (ii) the issuance of an aggregate of 8,226 shares of Common Stock to a consultant pursuant to the terms of a consulting agreement between July 1, 2023 and September 1, 2023; (iii) the addition of 16,605 shares of restricted Common Stock that were issued but unvested prior to June 30, 2023 and which have subsequently vested; and (iv) the consummation of the Acquisition including the issuance of 564,972 shares of restricted Common Stock Common Stock to the Joyce Trust pursuant to the terms of the Asset Purchase Agreement, the payment of $2,500,000 cash consideration to the Joyce Trust and the assets acquired including cash and the liabilities assumed in Acquisition.
After giving effect to: (i) the pro forma adjustments set forth above; and (ii) the sale and issuance by us of 3,086,419 Units in this Offering, based on an assumed Offering Price of $1.62 per Unit and after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us, assuming the underwriter’s option is not exercised, our pro forma, as adjusted net tangible book value as of June 30, 2023 would have been $11.1 million, or $0.90 per share. This represents an immediate increase in pro forma net tangible book value of $0.14 per share to our existing stockholders and immediate dilution of $0.72 per share to investors purchasing Units in this Offering at the assumed Offering Price. The following table illustrates this dilution on a per share of common stock basis assuming the underwriters do not exercise their option to purchase additional shares of Common Stock:
| Assumed public offering price per Unit | $ | 1.62 | ||||||
| Net tangible book value per share as of June 30, 2023 | $ | 0.95 | ||||||
| Pro forma net tangible book value per share | $ | 0.77 | ||||||
| Increase in net tangible book value per share after giving effect to this Offering | $ | 0.13 | ||||||
| Pro forma, as adjusted, net tangible book value per share as of June 30, 2023, after giving effect to the offering | $ | 0.90 | ||||||
| Dilution per share to new investors in the offering | $ | 0.72 |
The dilution information discussed above is illustrative only and may change based on the actual initial public offering price and other terms of this offering.
Each $0.25 increase or decrease in the assumed Offering Price of $1.62 per Unit would increase or decrease, as applicable, our pro forma as adjusted net tangible book value per share to new investors by approximately $0.06, and would increase or decrease, as applicable, dilution per share to new investors in this Offering by approximately $0.06, assuming that the number of Units offered by us, as set forth on the cover page of this prospectus, remains the same and after deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us. If the underwriters exercise their option to purchase additional shares of Common Stock from us in full, the pro forma as adjusted net tangible book value per share of our Common Stock immediately after this Offering would be $0.92 per share, and the dilution in pro forma net tangible book value per share to new investors in this Offering would be $0.70 per share.
We may also increase or decrease the number of Units we are offering. A 250,000 Unit increase in the number of Units offered by us, as set forth on the cover page of this prospectus, would increase the pro forma as adjusted net tangible book value per share by $0.01 and decrease the dilution per share to investors participating in this Offering by $0.01, assuming the assumed Offering Price of $1.62 per Unit remains the same and after deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us. A 250,000 Unit decrease in the number of Units offered by us, as set forth on the cover page of this prospectus, would decrease the pro forma as adjusted net tangible book value per share after this Offering by $0.01 and increase the dilution per share to new investors participating in this Offering by $0.01 assuming the assumed Offering Price of $1.62 per Unit remains the same and after deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us.
The following table presents, on a pro forma, as adjusted basis as of June 30, 2023, after giving effect to the new investors purchasing Units in this Offering with respect to the number of shares purchased from us, the total consideration paid or to be paid to us, which includes net proceeds received from the issuance of Units, and the average price per Unit paid or to be paid to us at an assumed Offering Price of $1.62 per Unit before deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us:
| Shares Purchased | Total Consideration |
Average Price Per |
||||||||||||||||||
| Number | Percent | Amount | Percent | Share | ||||||||||||||||
| (in thousands) | ||||||||||||||||||||
| Existing stockholders | 9,216,883 | 75 | % | $ | 15,570,479 | 76 | % | $ | 1.69 | |||||||||||
| New investors |
3,086,419 |
25 | % | $ | 5,000,000 | 24 | % | $ | 1.62 | |||||||||||
| Totals |
12,303,302 |
100 | % | $ | 20,570,479 | 100 | % | $ | 1.67 | |||||||||||
| 46 |
The table below assumes the underwriters’ exercise their over-allotment option in full:
| Shares Purchased | Total Consideration |
Average Price Per |
||||||||||||||||||
| Number | Percent | Amount | Percent | Share | ||||||||||||||||
| (in thousands) | ||||||||||||||||||||
| Existing stockholders | 9,216,883 | 72 | % | $ | 15,570,479 | 73 | % | $ | 1.69 | |||||||||||
| New investors |
3,549,381 |
28 |
% | $ | 5,750,000 | 27 | % | $ | 1.62 | |||||||||||
| Totals |
12,766,264 |
100 |
% | $ | 21,320,479 | 100 | % | $ | 1.67 | |||||||||||
The number of shares of Common Stock to be outstanding before and after this Offering as reflected in the tables and discussion above in this “Dilution” section are based on 8,555,365 shares of Common Stock outstanding as of June 30, 2023, which does not include 125,815 shares of unvested restricted Common Stock as of such date, 16,605 of which have vested as of September 20, 2023, 9,216,883 shares of Common Stock outstanding on a pro forma basis as of the date of this prospectus; and 12,766,264 shares of Common Stock outstanding on a pro forma, as adjusted basis after giving effect to the sale and issuance by us of 3,592,814 Units in this Offering, and excludes:
| ● |
133,414 unvested shares of restricted Common Stock as of September 20, 2023, which are outstanding but subject to forfeiture and therefore not included in our balance sheet; |
|
| ● | 3,086,419 shares of Common Stock issuable upon the exercise of the Warrants underlying the Units sold in this Offering; | |
| ● | 462,962 shares of Common Stock issuable upon the exercise of the Over-Allotment Option; | |
| ● | 462,962 shares of Common Stock issuable upon the exercise of 462,962 Warrants issuable upon the exercise of the Over-Allotment Option; | |
| ● | 61,728 shares of Common Stock issuable upon the exercise of the Representative’s Warrant; | |
| ● | 806,392 shares of Common Stock issuable on the exercise of stock options issued under our 2014 Equity Incentive Plan to certain of our employees, directors, and consultants with a weighted average exercise price equal to $4.33; | |
| ● | an aggregate of 4,305,812 shares of Common Stock issuable upon the exercise of outstanding Tradeable Warrants and Non-Tradeable Warrants (which share number reflects an adjustment in the number of shares to be issued upon exercise of the Tradeable Warrants and Non-Tradeable Warrants that will be effected upon consummation of this Offering in accordance with the price protection provisions contained in the Tradeable Warrants and Non-Tradeable Warrants) all of which, upon consummation of the Acquisition, will have an exercise price that will be reduced to $3.0625 per share; | |
| ● | 2,900,904 shares of Common Stock issuable on the exercise of outstanding warrants issued prior to consummation of our initial public offering with a weighted average exercise price equal to $5.31 per share; and | |
| ● | 658,294 shares of our Common Stock that are reserved for equity awards that may be granted under our 2014 Equity Incentive Plan. |
| 47 |
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITIONS AND RESULTS OF OPERATIONS
You should read the following discussion and analysis of our financial condition and results of operations together with our financial statements and related notes appearing at the end of this prospectus. Some of the information contained in this discussion and analysis is set forth at the end of this prospectus, including information with respect to our plans and strategy for our business and related financing, includes forward-looking statements that involve risks and uncertainties. As a result of many factors, including those factors set forth in the section entitled “Risk Factors,” our actual results could differ materially from the results described in or implied by the forward-looking statements contained in the following discussion and analysis. You should carefully read the section entitled “Risk Factors” to gain an understanding of the important factors that could cause actual results to differ materially from our forward- looking statements. Please also see the section entitled “Cautionary Note Regarding Forward-Looking Statements” and “Market, Industry and Other Data.”
Overview
This section presents management’s perspective on our financial condition and results of operations. The following discussion and analysis is intended to highlight and supplement data and information presented elsewhere in this prospectus, and should be read in conjunction with our interim unaudited condensed consolidated financial statements and notes included in this prospectus and our audited consolidated financial statements and the related notes and the discussion under the heading “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” included in this prospectus. The MD&A is also intended to provide you with information that will assist you in understanding our consolidated financial statements, the changes in key items in those consolidated financial statements from year to year, and the primary factors that accounted for those changes. To the extent that this discussion describes prior performance, the descriptions relate only to the periods listed, which may not be indicative of our future financial outcomes. In addition to historical information, this discussion contains forward-looking statements that involve risks, uncertainties, and assumptions that could cause our financial results to differ materially from management’s expectations. Factors that could cause such differences are discussed in the “Cautionary Note Regarding Forward-Looking Statements” section of this prospectus and in the “Risk Factors” section of this prospectus.
Data as of and for the six months ended June 30, 2023 and 2022, has been derived from our unaudited condensed consolidated financial statements appearing in this prospectus and data as of and for the year ended December 31, 2022 and 2021 has been derived from our audited condensed consolidated financial statements appearing in this prospectus. Results for any interim period should not be construed as an inference of what our results would be for any full fiscal year or future period.
Our MD&A is organized as follows:
| ● | Company Overview – Discussion of our business plan and strategy to provide context for the remainder of the MD&A. | |
| ● | Results of Operations – Analysis of our financial results comparing (i) the six months ended June 30, 2023, to the comparable period in 2022 and (ii) the year ended December 31, 2022 to the comparable period in 2021. | |
| ● | Liquidity and Capital Resources – Analysis of changes in our cash flows, and discussion of our financial condition and potential sources of liquidity. | |
| ● | Critical Accounting Estimates – Accounting policies that we believe are important to understanding the assumptions and judgments incorporated in our reported financial results and forecasts. |
Company Overview
Business
We develop noninvasive diagnostics to detect early-stage lung cancer and other diseases of the lung. We are developing our platform technologies so that, in the future, they could result in broad-spectrum cancer treatments. We develop proprietary noninvasive diagnostic tests using technology that preferentially targets cancer cells and cell populations indicative of a diseased state. Research and optimization of our platform technologies are conducted in our laboratories at The University of Texas at San Antonio.
Our first diagnostic test, CyPath® Lung, addresses the need for noninvasive detection of early-stage lung cancer. Lung cancer is the leading cause of cancer-related deaths. Physicians will be able to order CyPath® Lung to assist in their assessment of patients who are at high risk for lung cancer. The CyPath® Lung test enables physicians to more confidently identify patients who will likely benefit from timely intervention and more invasive follow-up procedures and is another tool to help distinguish them from patients who are likely without lung cancer and should continue annual screening. CyPath® Lung has the potential to increase overall diagnostic accuracy of lung cancer, which could lead to increased survival, fewer unnecessary invasive procedures, reduced patient anxiety, and lower medical costs.
Through our wholly owned subsidiary, OncoSelect®, our research has led to discoveries of novel potential cancer therapeutics that specifically and selectively target cancer cells that have been grown in petri dishes.
| 48 |
Recent Developments
On September 18, 2023, PPLS consummated the Acquisition of the Laboratory Assets pursuant to the terms of the Asset Purchase Agreement with Village Oaks and Dr. Joyce. As a result of the Acquisition, the CAP-accredited CLIA-certified clinical pathology laboratory is owned by PPLS. Dr. Joyce was the Medical Director and Laboratory Director of the clinical pathology laboratory prior to the Acquisition and he continues to serve as Medical Director and Laboratory Director after the Acquisition. Founded in 2007 by Dr. Roby Joyce, Village Oaks, under the trade name Precision Pathology Services, has provided pathology services to physicians practicing in a variety of outpatient settings. Since September 2021, Village Oaks, under the trade name Precision Pathology Services, has offered CyPath® Lung for sale as an LDT for the detection of early-stage lung cancer. In addition to CyPath® Lung, PPLS intends to continue to offer a range of laboratory services including respiratory testing for SARS-CoV-2 and influenza, anatomical pathology, morphological stains, histological services, DNA extractions, STI testing and women’s and men’s health testing.
Pursuant to the terms of the Asset Purchase Agreement, PPLS acquired all of the Laboratory Assets, which included all of the assets owned by Village Oaks, other than medical assets, which are assets Village Oaks used in connection with its management and operation of a clinical pathology laboratory, now owned by PPLS, and related services business, and assumed certain liabilities and obligations. The laboratory is a clinical pathology laboratory that is CAP-accredited and CLIA-certified. Pursuant to the terms of the Asset Purchase Agreement, Village Oaks received $3,500,000 in consideration for the assets to be purchased by PPLS, of which $1,000,000 was paid by the issuance of 564,972 shares of our restricted Common Stock to the Joyce Trust, which share number was determined by dividing $1,000,000 by $1.77, the average of the trading day closing prices for the 30 days prior to September 15, 2023, rounded to the nearest whole share.
The Asset Purchase Agreement contains customary representations, warranties and covenants made by PPLS and Village Oaks and consummation of the transaction was subject to customary closing conditions, including, among other things, entry into the other ancillary agreements described below.
Pursuant to the Asset Purchase Agreement, PPLS assumed all liabilities and obligations under and obtained any and all rights, title and interest of Village Oaks in and to (i) the Assumed Leases pursuant to the Assumption Agreement; (ii) the Assumed Contracts under the Assumption Agreement; (iii) all accounts payable of Village Oaks as of September 18, 2023 that were incurred in the ordinary course of business consistent with past custom and practice; and (iv) the lease of the premises used in connection with operation of the CLIA-certified and CAP-accredited clinical pathology laboratory pursuant to the Assignment and Assumption of Lease, which Assignment of Lease was consented to by the landlord of the leased premises. The monthly rent is currently $10,143.83 per month and the term of the Lease is five years.
In connection with the Asset Purchase Agreement, PPLS entered into the Management Services Agreement with Village Oaks, pursuant to which PPLS will provide comprehensive management and administrative services to Village Oaks in connection with the operation of Village Oaks’ professional cytopathology, histopathology, clinical and anatomic pathology interpretation medical services practice. PPLS will provide space, equipment, administrative, management and clinical personnel, billing and collection, and related management services to Village Oaks in exchange for a management fee of 70% of the net revenues received by Village Oaks from the provision of the medical services. The Management Services Agreement has an initial term of 20 years and provides that upon expiration of the initial term, it will be automatically extended for two additional successive terms of five years each, unless either party delivers written notice of its intention not to extend the term of the agreement not less than 90 days prior to the expiration of the preceding term. The Management Services Agreement also provides that until the fifth anniversary of its effective date, Village Oaks will not, without the prior written approval of PPLS own, operate or have any financial interest in any other person or entity that operates an independent laboratory or an enterprise within the United States that provides or promotes management or administrative services or any product or services substantially similar to those provided by PPLS.
In connection with the Asset Purchase Agreement, PPLS entered into the Succession Agreement, pursuant to which Dr. Joyce, as holder of 100% of the issued and outstanding stock of Village Oaks, and Village Oaks are restricted from disposing of their equity interests in Village Oaks, subject to certain exceptions, without the prior written consent of us and Village Oaks. The Succession Agreement further provides that the entire equity interest held by Dr. Joyce in Village Oaks will be automatically assigned and transferred to a successor who meets the Eligibility Requirements of a Designated Physician, as such terms are defined and described in the Succession Agreement, in the event of, among other things, the death, disability, retirement, or a court’s determination of incompetence of Dr. Joyce, as well as Dr. Joyce’s failure to satisfy the eligibility requirements of a Designated Physician, exclusion or disqualification from participation in the Medicare program, conviction of a felony or crime or moral turpitude, bankruptcy filing, or material breach of the Succession Agreement. In the event of the automatic transfer of Dr. Joyce’s equity interests in Village Oaks as provided in the Succession Agreement, such agreement provides that the board of directors of Village Oaks shall nominate a group of three candidates as the Designated Physician who satisfy the Eligibility Requirements. In the event the Company desires not to select any of such candidates, the Company shall select and appoint a successor Designated Physician from any other physicians that satisfy the Eligibility Requirements. Subject in all cases to the Management Services Agreement, Dr. Joyce shall not cause any voluntary interruption of the conduct of Village Oaks’ business and operations, and shall use commercially reasonable efforts to preserve (or assist us in preserving) all rights, privileges and franchises held by Village Oaks, including the maintenance of all contracts, copyrights, trademarks, licenses and registrations.
| 49 |
In connection with the Asset Purchase Agreement, PPLS entered into the Professional Services Agreement with Village Oaks, pursuant to which Village Oaks will provide pathology interpretation services as requested on behalf of PPLS based on the professional fees approved for the CPT code for the services provided under the Medicare Physician Fee Schedule in the locality where the test is performed. The Professional Services Agreement has an initial term of 20 years and provides that upon expiration of the initial term, it will be automatically extended for successive terms of twelve months each, unless either party delivers written notice of its intention not to extend the term of the agreement not less than 30 days prior to the expiration of the preceding term.
In connection with the Asset Purchase Agreement, we entered into the Executive Employment Agreement with Dr. Joyce, for a term of three years, to serve as the Medical Director and Laboratory Director of PPLS at a base salary of $333,333.34 per year. Pursuant to the Joyce Employment Agreement, Dr. Joyce was also appointed to serve on our Board of Directors. Dr. Joyce will be eligible to participate in or receive benefits under our benefit plans generally made available to executives of similar status and responsibilities and will be provided use of a company car. In the event the Joyce Employment Agreement is terminated for any reason, including by Dr. Joyce upon 60 days’ notice, by us for cause or by reason of Dr. Joyce’s death, Dr. Joyce (or his estate as applicable) will receive his base salary for the remainder of the three-year employment term. However, the Joyce Employment Agreement provides that if Dr. Joyce breaches any of the restrictive covenants set forth in the Joyce Employment Agreement, including a covenant not to compete during his term of employment and a covenant not to knowingly disclose confidential information, such breach will be grounds for the immediate termination of Dr. Joyce and will result in the forfeiture of all compensation and benefits otherwise due to Dr. Joyce.
One of the Assumed Leases is the Hologic Equipment Lease, pursuant to which PPLS leases reagent equipment from Hologic and is required to purchase a minimum number of specified testing kits each year. The total monthly minimum purchase commitment PPLS is required to pay Hologic, inclusive of the lease of the reagent equipment, is $16,914 per month. The term of the Hologic Equipment Lease currently expires on December 20, 2027.
Another of the Assumed Leases is the Leica Equipment Lease, pursuant to which PPLS leases reagent equipment from Leica and is required to purchase a minimum number of specified testing kits. The total monthly minimum purchase commitment PPLS is required to pay to Leica, inclusive of the lease of the reagent equipment, is $19,790 per month. The term of the Leica Equipment Lease currently expires on March 23, 2026.
One of the Assumed Contracts is the License Agreement. Pursuant to the License Agreement, Pathology Watch granted a license to its digital imaging cloud-based pathology platform to facilitate remote interpretation and billing of pathology specimens by qualified professionals to PPLS for a monthly fee of $25,000. In connection with the License Agreement, Pathology Watch also provides certain support services and marketing vendor services to PPLS for the monthly fee of $38,000, for a total monthly fee paid by PPLS to Precision Watch of $63,000. The License Agreement is for an initial term of twelve months, unless terminated by either party upon 90 days’ notice, and provides that upon expiration of the initial term (or any renewal term), it will be automatically extended for successive twelve month terms, unless either party notifies the other party of its intention not to renew the License Agreement not less than 90 days prior to the expiration of the current term.
In connection with the Asset Purchase Agreement, Dr. Joyce, on behalf of Village Oaks, executed the Bill of Sale, pursuant to which all rights, title, and interest of Village Oaks in and to the permits listed on Exhibit A attached thereto, inclusive of the CLIA-certificate and CAP-accreditation, notwithstanding the transfer of the CLIA certificate by operation of law to PPLS upon consummation of the Acquisition, were confirmed to have been transferred and assigned to PPLS.
Amendment to Warrants
On September 17, 2023, Mr. Girgenti, the Cranye Girgenti Testamentary Trust, Gary Rubin, The Harvey Sandler Revocable Trust, a trust of which Mr. Rubin is a co-trustee, Ms. Zannes and Dr. Joyce consented to an amendment of the terms of the outstanding warrants that they own. Such warrants include warrants (i) Tradeable Warrants to purchase 98,198, 39,182, and 39,182 shares of Common Stock owned by Mr. Girgenti, The Harvey Sandler Revocable Trust, and Ms. Zannes, respectively); (ii) Non-Tradeable Warrants to purchase 102,286, 40,813, and 40,813 shares of Common Stock owned by Mr. Girgenti, The Harvey Sandler Revocable Trust, and Ms. Zannes, respectively; and (iii) Pre-IPO Warrants to purchase 469,063, 8,332, 571,373, 23,571, 17,137, and 14,285 shares of Common Stock owned by Mr. Girgenti, the Cranye Girgenti Testamentary Trust, Mr. Rubin, The Harvey Sandler Revocable Trust, Ms. Zannes and Dr. Joyce, respectively. The Warrant Amendment provides that such warrants will not be exercisable until the date that we file a certificate of amendment to our certificate of incorporation with the State of Delaware which increases the number of shares of our authorized Common Stock to allow for sufficient authorized and unissued shares of Common Stock for the full exercise of all of the outstanding Pre-IPO Warrants, Tradeable Warrants and Non-Tradeable Warrants of the Company and the issuance of all of the shares of Common Stock underlying such warrants.
Financial
To date, we have devoted a substantial portion of our efforts and financial resources to the development of our first diagnostic test, CyPath® Lung. Since our inception in 2014, we have funded our operations principally through the proceeds of our initial public offering and private sales of our equity or debt securities. On September 6, 2022, we completed our initial public offering of our securities pursuant to which we raised gross proceeds of $7.9 million. As of September 20, 2023, investors participating in the initial public offering exercised a total of 725,576 Tradeable Warrants at a price of $7.35 per share and 310,910 Non-Tradable Warrants at a price of $7.656 per share. Combined with our underwritten public offering, we received an aggregate of approximately $15.6 million as of September 28, 2022. We believe that our available cash together with the proceeds of this Offering will be sufficient to fund our planned operations for at least 12 months following the date of this prospectus.
| 50 |
In the second quarter of 2022, we started to recognize revenue from sales of the CyPath® Lung test by Village Oaks, to which we had previously granted a license to develop CyPath® Lung for commercialization and to manufacture, use, market and sell CyPath® Lung as an LDT prior to the Acquisition, which license was assigned to and assumed by PPLS in connection with the Acquisition. We have never been profitable, and as of June 30, 2023, we had total working capital of $7.9 million and an accumulated deficit of approximately $39.9 million. As of June 30, 2023, we had cash and cash equivalents of $8.3 million. We expect to continue to incur significant operating losses for the foreseeable future as we continue the development of our diagnostic tests and therapeutic products and advance our diagnostic tests through clinical trials.
We anticipate raising additional cash needed through the private or public sales of equity or debt securities, collaborative arrangements, or a combination thereof, to continue to fund our operations and develop our products. There is no assurance that any such collaborative arrangement will be entered into or that financing will be available to us when needed in order to allow us to continue our operations, or if available, on terms acceptable to us. If we do not raise sufficient funds in a timely manner, we may be forced to curtail operations, delay our clinical trials, cease operations altogether, or file for bankruptcy.
Results of Operations
Six Months Ended June 30, 2023, Compared to Six Months Ended June 30, 2022
Net loss for the six months ended June 30, 2023, was approximately $3.2 million, compared to a net loss of approximately $1.6 million for the six months ended June 30, 2022, resulting from the operational activities described below and a prior year non-cash FMV adjustment of $1.2 million relating to convertible notes. The increase in net loss was primarily attributed to a prior year non-cash adjustment of $1.6 million related to the fair value of convertible notes that were outstanding during the quarter ended June 30,2022 and were no longer outstanding during the quarter ended June 30, 2023.
Revenue
To date, revenue has been generated in three ways: (1) royalties from our diagnostic test, CyPath® Lung , (2) clinical flow cytometry services provided to Village Oaks related to our CyPath® Lung test, and (3) CyPath® Lung tests purchased by the DOD for an observational study, “Detection of Abnormal Respiratory Cell Populations in Lung Cancer Screening Patients Using the CyPath® Lung Assay (NCT05870592),” and research and development on using bronchoalveolar lavage fluid as a biological sample to assess cardiopulmonary function and exercise performance in military personnel post COVID-19 infection. Village Oaks, to which we had previously granted a license to develop CyPath® Lung for commercialization and to manufacture, use, market and sell CyPath® Lung as an LDT prior to the Acquisition, which license was assigned to and assumed by PPLS in connection with the Acquisition, began a limited market launch in the second quarter of 2022 to pulmonologists, internists, and general practitioners in the South Texas area designed to refine future positioning and develop strategic marketing insight for our CyPath® Lung test, which resulted in royalties revenue. The services are completed upon release of a patient’s test result to the ordering healthcare provider. Upon consolidation of our financial statements with PPLS, we will be eliminating the revenue from clinical flow cytometry services that were previously provided to Village Oaks because, upon consummation of the Acquisition, such services are provided to PPLS, our wholly owned subsidiary.
In the first quarter of 2023, we engaged the marketing and advertising firms of Havas Health & You and Trinity Life Sciences to build the CyPath® Lung brand and position it for success in the cancer diagnostics sector. Havas Health & You, a large global health network, is creating the branding to align with the need for a patient-friendly diagnostic that gives physicians another tool to assess the potential or presence of lung cancer in their high-risk patients. Trinity Life Sciences is using the insights and analytics it has gathered from healthcare practitioners to focus the short-term objectives of our marketing strategy for CyPath® Lung. The limited test market launch in South Texas is designed to evaluate our marketing program and help us ensure each step in the care pathway – from the initial order by physicians to sputum collection and processing, to generating and delivering the patient report – is efficient and effective. This limited test market approach allows us to refine future positioning and develop strategic insight for our CyPath® Lung test before expanding to a larger market.
In addition to introducing pulmonologists, family practitioners, and other providers to CyPath® Lung, we are selling CyPath® Lung tests to the DOD for an observational study, “Detection of Abnormal Respiratory Cell Populations in Lung Cancer Screening Patients Using the CyPath® Lung Assay,” and for research and development on using bronchoalveolar lavage fluid as a biological sample to assess cardiopulmonary function and exercise performance in military personnel post COVID-19 infection. The DOD represents a significant potential market for CyPath® Lung. Including sales to the DOD, clinical services provided to Village Oaks, and direct sales of CyPath® Lung as an LDT to physicians, we had revenue of approximately $21,000 during the six months ended June 30, 2023, compared to no revenue in 2022. Sales to the DOD represented $10,000 (48%), royalty income represented $8,000 (38%) and services represented $3,000 (14%) of total revenues.
| 51 |
We expect revenues from CyPath® Lung to continue to grow as we add physicians prescribing our diagnostic test and expand our outreach to other geographic areas and larger medical systems. Our revenues are affected by the test volume of our products, patient adherence rates, payer mix, the levels of reimbursement, and payment patterns of payors and patients.
|
Six Months Ended June 30,(1) |
Change in 2023 Versus 2022 |
|||||||||||||||
| 2023 | 2022 | $ | % | |||||||||||||
| (amount in thousands) | (unaudited) | |||||||||||||||
| Revenue | ||||||||||||||||
| Royalty income (CyPath® Lung sales) | $ | 8 | $ | 1 | $ | 7 | 493 | % | ||||||||
| Clinical services (flow cytometry) | 3 | — | 3 | 100 | % | |||||||||||
| DoD observational study (CyPath® Lung) | 10 | — | 10 | 100 | % | |||||||||||
| Total revenue | $ | 21 | $ | 1 | $ | 20 | 1566 | % | ||||||||
Cost of Sales
Cost of sales is comprised primarily of costs related to inventory production and usage and shipment of collection kits to patients and healthcare providers. The increase in cost of sales for the six months ended June 30, 2023, is primarily due to sales of our diagnostic kits during the quarter, compared to no sales in the prior year.
Operating Expenses
|
Six Months Ended June 30,(1) |
Change in 2023 Versus 2022 |
|||||||||||||||
| 2023 | 2022 | $ | % | |||||||||||||
| (amount in thousands) | (unaudited) | |||||||||||||||
| Operating expenses | ||||||||||||||||
| Research and development | $ | 705 | $ | 528 | $ | 176 | 33 | % | ||||||||
| Clinical development | 55 | 81 | (26 | ) | -32 | % | ||||||||||
| Selling, general and administrative | 2,596 | 803 | 1,793 | 223 | % | |||||||||||
| Total operating expenses | $ | 3,356 | $ | 1,412 | $ | 1,943 | 138 | % | ||||||||
| (1) | Represents operating expenses from our unaudited condensed consolidated financial statements for the six-month period ended June 30, 2023 and 2022, respectively. |
Operating expenses totaled approximately $3.4 million and $1.4 million during the six months ended June 30, 2023 and 2022, respectively. The increase in operating expenses is the result of the following factors.
Research and Development Expenses
Our research and development expenses consist primarily of expenditures for lab operations, clinical and preclinical studies, compensation, and consulting costs.
Research and development expenses totaled approximately $705,000 and $528,000 for the six months ended June 30, 2023 and 2022, respectively. The increase of approximately $176,000, or 33%, for the six months ended June 30, 2023, compared to the same period in 2022, was primarily due to an increase in compensation costs and benefits due to additional research personnel, as well as a related increase in costs for lab supplies and reagents. Additionally, equipment costs, including depreciation and maintenance costs, increased as we purchased capital equipment to support research and development efforts.
Clinical Development
Clinical development expenses totaled approximately $55,000 and $81,000 for the six months ended June 30, 2023 and 2022, respectively. The decrease of approximately $26,000, or 32%, for the six months ended June 30, 2023, compared to the same period in 2022, was primarily attributable to a decrease in professional fees, including consulting fees, related to evaluating the clinical strategy in the prior year for our pivotal clinical trial designed to confirm the sensitivity and specificity of CyPath® Lung in detecting lung cancer in persons at high risk for the disease, including patients who display indeterminate lung nodules between 6mm and 30mm in size which often present a challenge in diagnosis.
| 52 |
Selling, General and Administrative
Our selling, general and administrative expenses consist primarily of expenditures related to employee compensation, legal, accounting and tax, other professional services, and general operating expenses.
Selling, general and administrative expenses totaled approximately $2.6 million and $803,000 for the six months ended June 30, 2023 and 2022, respectively. The increase of approximately $1.8 million, or 223%, for the six months ended June 30, 2023, compared to the same period in 2022, was primarily attributable to an increase in consulting, legal, and professional fees incurred in 2023 compared to 2022 to comply with the reporting requirements of a public company, as well as an increase related to board compensation. Patent costs increased in the current year as we maintain and expand our patent portfolio to protect our diagnostic and therapeutic platforms. Additionally, compensation increased due to additional personnel and support services to support the launch of sales of our diagnostic test, CyPath® Lung.
Other Income (Expense)
| Six Months Ended | Change in 2023 | |||||||||||||||
| June 30, | Versus 2022 | |||||||||||||||
| 2023 | 2022 | $ | % | |||||||||||||
| (amount in thousands | (unaudited) | |||||||||||||||
| Interest income (expense), net | $ | 80 | $ | (1,546 | ) | $ | 1,626 | 105 | % | |||||||
| Gain (loss) on change in fair value of convertible notes | — | 1,399 | (1,399 | ) | -100 | % | ||||||||||
| Total other income (expense) | $ | 80 | $ | (148 | ) | $ | 228 | 154 | % | |||||||
Other income (expense), net totaled approximately $80,000 and ($148,000) for the six-month period ended June 30, 2023 and 2022, respectively.
Interest Income (Expense), net
Interest income (expense), net was approximately $80,000 for the six months ended June 30, 2023, compared to ($1.5) million for the six months ended June 30, 2022. The change was due to no convertible notes outstanding during the current year compared to the same period in the prior year, as all convertible and bridge notes were converted as a result of our IPO in the prior year. Additionally, in 2022 we recorded interest expense for the amortization of debt discount related to the issuance of bridge notes.
Gain (loss) on change in fair value of convertible notes
There was a gain of approximately $1.4 million on the change in fair value of convertible notes during the six months ended June 30, 2022, compared to no loss during the six months ended June 30, 2023. The change in the fair value of convertible notes resulted primarily from changes in the calculation of the fair value of our stock, the reduction in the expected term, and other assumptions during the reported periods. All convertible and bridge notes were converted as a result of our initial public offering in the prior year, resulting in no additional changes in fair value related to the convertible and bridge notes.
Year Ended December 31, 2022, Compared to the Year Ended December 31, 2021
Our results of operations have varied significantly from year to year and quarter to quarter and may vary significantly in the future. Net loss for the year ended December 31, 2022, was approximately $8.2 million, compared to a net loss of approximately $6.3 million for the year ended December 31, 2021, resulting from the operational activities described below.
Revenue
Our revenue during the year ended December 31, 2022 was generated exclusively from royalties for our first diagnostic test, CyPath® Lung, from sales by Village Oaks, a CAP-accredited, CLIA-certified clinical pathology laboratory to which we had previously granted a license to develop CyPath® Lung for commercialization and to manufacture, use, market and sell CyPath® Lung as an LDT prior to the Acquisition, which license was assigned to and assumed by PPLS in connection with the Acquisition. Although Village Oaks placed CyPath® Lung on its list of tests offered to physicians in second quarter 2022, there was limited marketing of the product until our initial public offering that was consummated in September provided funds to assemble a marketing team of experts focused on demonstrating the clinical value of CyPath® Lung in the marketplace. The limited test market launch in the San Antonio, Texas area is designed to evaluate our marketing program and help us ensure each step in the care pathway is efficient and effective, from the initial order by physicians to patient sputum collection and processing, to generating and delivering the patient report. This limited test market approach allows us to refine future positioning and develop strategic insight for our CyPath® Lung test before expanding to a larger market. We had revenue of approximately $5,000 during the year ended December 31, 2022, from the sale of CyPath® Lung as an LDT, compared to no revenue in 2021.
We expect our revenue to grow for CyPath® Lung as we add physicians prescribing our diagnostic test and expand our outreach to other geographic areas. Our revenues are affected by the test volume of our products, patient adherence rates, payer mix, the levels of reimbursement, and payment patterns of payors and patients.
| 53 |
Cost of Sales
Cost of sales is comprised primarily of costs related to inventory production and usage and shipment of collection kits to patients and healthcare providers. The increase in cost of sales for the year ended December 31, 2022, is primarily due to the launch of sales in the second quarter of 2022, compared to no sales in the prior year.
Operating Expenses
| Year Ended | Change in 2022 | |||||||||||||||
| December 31, | Versus 2021 | |||||||||||||||
| 2022 | 2021 | $ | % | |||||||||||||
| (amounts in thousands) |
(amounts in thousands) |
|||||||||||||||
| Operating expenses: | ||||||||||||||||
| Research and development | $ | 1,143 | $ | 1,008 | $ | 135 | 13 | % | ||||||||
| Clinical development | 146 | 130 | 16 | 12 | % | |||||||||||
| Selling, general and administrative | 2,727 | 1,069 | 1,658 | 155 | % | |||||||||||
| Total operating expense | $ | 4,016 | $ | 2,207 | $ | 1,809 | 82 | % | ||||||||
Operating expenses totaled $4.0 million and $2.2 million during 2022 and 2021, respectively. The increase in operating expenses is the result of the following factors.
Research and Development
Our research and development expenses consist primarily of expenditures for lab operations, preclinical studies, compensation, and consulting costs.
Research and development expenses totaled $1.1 million and $1.0 million during the year ended December 31, 2022 and 2021, respectively. The increase of approximately $135,000, or 13%, for the year ended December 31, 2022 compared to 2021 was primarily attributable to an increase in compensation costs and benefits as we added research personnel, partially offset by a decrease in the prior year due to several employees who were furloughed for several months and later returned to their positions with us. Additionally, costs related to lab supplies and reagents increased as employees returned to facilities after restrictions eased for COVID-19. These increases were partially offset by a decrease in stock compensation expense related to stock option and restricted share grants to employees and consultants in 2022 compared to 2021.
Clinical Development
Clinical development expenses totaled approximately $146,000 and $130,000 during the year ended December 31, 2022 and 2021, respectively. The increase of approximately $16,000, or 12%, for 2022, compared to 2021 was primarily attributable to an increase in professional fees, including consulting fees, and increases in clinical study activities related to site costs compared to 2021 as operations were still being affected by the global pandemic.
Selling, General and Administrative
Our selling, general and administrative expenses consist primarily of expenditures related to compensation, legal, accounting, tax and other professional services, and general operating costs.
Selling, general and administrative expenses totaled approximately $2.7 million and $1.1 million during the year ended December 31, 2022 and 2021, respectively. The increase of $1.6 million, or 155%, for the year ended December 31, 2022 compared to the year ended December 31, 2021 was primarily attributable to increases related to consulting, legal, and professional fees incurred in 2022 compared to 2021 as we prepared for our initial public offering and comply with the reporting requirements of a public company. Patent costs increased in the current year as we maintain and expand our patent portfolio to protect our diagnostic and therapeutic platforms. Additionally, the increase was due to an increase in stock-based compensation, as well as an increase in compensation and benefits as we increased personnel and support services to support the launch of sales of our diagnostic test, CyPath® Lung.
Other Income (Expense)
| Year Ended | Change in 2022 | |||||||||||||||
| December 31, | Versus 2021 | |||||||||||||||
| 2022 | 2021 | $ | % | |||||||||||||
| (amounts in thousands) |
(amounts in thousands) |
|||||||||||||||
| Interest income (expense), net | $ | (2,486 | ) | $ | (1,002 | ) | $ | (1,484 | ) | 148 | % | |||||
| Gain on extinguishment of debt | 212 | 239 | (27 | ) | -11 | % | ||||||||||
| Fair value of warrants | — | (4,080 | ) | 4,080 | -100 | % | ||||||||||
| Loss on change in fair value of convertible notes | (1,867 | ) | 725 | (2,592 | ) | -358 | % | |||||||||
| Total other income (expense) | $ | (4,141 | ) | $ | (4,118 | ) | $ | (23 | ) | 1 | % | |||||
Other income (expense) totaled approximately ($4.1) million and ($4.1) million for the year ended December 31, 2022 and 2021, respectively.
| 54 |
Interest income (expense)
We had net interest expense of approximately $2.5 million and $1.0 million for the years ended December 31, 2022 and 2021, respectively. The increase was due to additional convertible notes outstanding during the same period in the prior year. Additionally, in 2022 we recorded interest expense of approximately $2.1 million for the amortization of debt discount related to the issuance of Bridge Notes.
Gain on Extinguishment of Debt
In March 2021, we received a second draw $0.2 million Paycheck Protection Program Loan (the “PPP Loan”) and received forgiveness from the Small Business Administration (SBA) in April 2022, recording a gain of $212,000 on the extinguishment of the PPP Loan. In April 2020, we received an initial $0.2 million PPP Loan and received forgiveness from the SBA in June 2021, recording a gain of $239,000 on the extinguishment of the PPP Loan.
Fair value of warrants
In 2021, in connection with the issuance of the Bridge Notes, we amended the terms of certain convertible notes. As an inducement to amending the notes, we issued Common Stock warrants with the same terms and conditions as the warrants issued to the Bridge Note holders. The estimated fair value of the warrants was $4.1 million and immediately expensed within the accompanying consolidated statement of operations. The Bridge Notes converted to Common Stock upon the consummation of the initial public offering.
(Loss) gain on change in fair value of convertible notes
The loss on the change in fair value of convertible notes totaled approximately $1.9 million during 2022 compared to a gain of $0.7 million during 2021, respectively. The change in the fair value of convertible notes resulted primarily from changes in the calculation of the fair value of our stock, the reduction in the expected term, and other assumptions during the reported periods. Refer to our notes to audited financial statements for further discussion on our convertible notes.
Liquidity and Capital Resources
Six Months Ended June 30, 2023, Compared to Six Months Ended June 30, 2022
To date, we have funded our operations primarily through our initial public offering, exercise of warrants, and the sale of our equity and debt securities, resulting in gross proceeds of approximately $34.3 million.
We have incurred losses since our inception in 2014 as a result of significant expenditures for operations and research and development and, prior to April 2022, the lack of any approved diagnostic test or therapeutic products to generate revenue. For the six months ended June 30, 2023 and 2022, we had net losses of $3.2 million and $1.6 million, respectively, and we expect to incur substantial additional losses in future periods. We have an accumulated deficit of approximately $39.9 million as of June 30, 2023 and approximately $36.7 million as of December 31, 2022. Cash and cash equivalents were approximately $8.3 million as of June 30, 2023 and approximately $11.4 million as of December 31, 2022. Based on our current level of expected operating expenditures and the proceeds from this Offering, we expect to be able to fund our operations for at least 12 months following the date of this prospectus.
As a result of the Acquisition, we anticipate that our revenue will increase. We also anticipate an increase in selling, general and administrative expenses due to the increase in expenses in operating the CAP/CLIA clinical pathology laboratory, including the expense related to additional employees.
We continue to seek sources of financing to fund our continued operations and research and development programs. To raise additional capital, we may sell additional equity or debt securities, or enter into collaborative, strategic, and/or licensing transactions. There can be no assurance that we will be able to complete any financing transaction in a timely manner or on acceptable terms or otherwise or enter into a collaborative or strategic transaction. If we are not able to raise additional cash, we may be forced to delay, curtail, or cease development of our diagnostic tests or therapeutic products, or cease operations altogether.
Summary Statements of Cash Flows
The following information reflects cash flows for the periods presented:
| Six Months Ended | ||||||||
| June 30, | ||||||||
| 2023 | 2022 | |||||||
| (amounts in thousands) | (unaudited) | |||||||
| Cash and cash equivalents at beginning of period | $ | 11,414 | $ | 1,361 | ||||
| Net cash used in operating activities | (2,889 | ) | (1,010 | ) | ||||
| Net cash used in investing activities | (36 | ) | — | |||||
| Net cash used in financing activities | (210 | ) | (136 | ) | ||||
| Cash and cash equivalents at end of period | $ | 8,279 | $ | 215 | ||||
Net Cash Used in Operating Activities
Net cash used in operating activities was approximately $2.9 million and $1.0 million for the six months ended June 30, 2023 and 2022, respectively. The increase of approximately $1.8 million in cash used by operations during the six months ended June 30, 2023, compared to the same period in 2022, was primarily attributable to an increase of $1.7 million in our loss from operations as compared to the prior year as described above.
Net Cash Used in Investing Activities
We used approximately $36,000 for the six months ended June 30, 2023, in investing activities related to the purchase of computer and lab equipment, compared to no cash used in investing activities for the six months ended June 30, 2022.
Net Cash Used by Financing Activities
Cash used in financing activities was approximately $209,000 compared to cash used in financing activities of approximately $136,000 for the six months ended June 30, 2023, and 2022, respectively. The change in cash used in financing activities for the six months ended June 30, 2023, compared to 2022, was a result of the short-term financing we obtained for director and officer insurance policies, compared to net proceeds from bridge notes of approximately $0.5 million in the same period in the prior year, offset by the payment of deferred issuance costs related to our initial public offering completed in September 2022.
| 55 |
Year Ended December 31, 2022, Compared to the Year Ended December 31, 2021
Cash Flows
The following information reflects cash flows for the years presented:
| Year Ended | ||||||||
| December 31, | ||||||||
| 2022 | 2021 | |||||||
| (amounts in thousands) | ||||||||
| Cash and cash equivalents at beginning of year | $ | 1,361 | $ | 83 | ||||
| Net cash used in operating activities | (4,071 | ) | (2,049 | ) | ||||
| Net cash used in investing activities | (220 | ) | — | |||||
| Net cash provided by financing activities | 14,344 | 3,327 | ||||||
| Cash and cash equivalents at end of year | $ | 11,414 | $ | 1,361 | ||||
Net Cash Used in Operating Activities
Net cash used in operating activities was approximately $4.1 million and $2.0 million for the years ended December 31, 2022 and 2021, respectively. The increase of approximately $2.1 million in cash used by operations during the year ended December 31, 2022, compared to 2021, was primarily attributable to an increase of $1.8 million in our loss from operations as compared to the prior year as described above. These increases were partially offset by adjustments for the amortization of debt discount related to the issuance of Bridge Notes and non-cash charges related to stock-based compensation.
Net Cash Used in Investing Activities
We used approximately $0.2 million in investing activities in 2022, compared to no cash used for the year ended December 31, 2021. The increase in cash used in investing activities in 2022, compared to 2021, is attributable to the purchase of lab equipment in the fourth quarter of 2022.
Net Cash Provided by Financing Activities
During the year ended December 31, 2022, net cash provided by financing activities was $14.3 million primarily due to net proceeds of approximately $6.0 million from issuance of Common Stock in our initial public offering, as well as proceeds of approximately $7.8 million from the exercise of warrants and options. Additionally, the increase is due to the issuance of $0.7 million of our Bridge Notes during the year, partially offset by debt issuance costs and the repayment of $425,000 of notes not converted as part of our initial public offering.
During the year ended December 31, 2021, net cash provided by financing activities was $3.3 million consisting of $3.3 million from the issuance of convertible notes, as well as receiving a second draw on our PPP Loan of $212,000 in March 2021, partially offset by the payment of approximately $180,000 in debt issuance costs. In April 2022, the Company submitted an application for forgiveness for the second draw on our PPP Loan and received notice of forgiveness from the SBA.
Contractual Obligations and Commitments
We enter into contracts in the normal course of business with third-party contract organizations for clinical trials, and other services and products used for research and development and operating purposes. These contracts generally provide for termination following a certain period after notice, and therefore we believe that any non-cancelable obligations under these agreements are not material.
Critical Accounting Estimates
The preparation of financial statements in conformity with GAAP requires management to make significant judgments and estimates that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Management bases these significant judgments and estimates on historical experience and other assumptions it believes to be reasonable based upon information presently available. Actual results could differ from those estimates under different assumptions, judgments or conditions.
| 56 |
Stock-Based Compensation
We follow ASC 718, Compensation – Stock Compensation, which requires the measurement and recognition of compensation expense for all share-based payment awards made to employees, directors, and non-employees based on estimated fair values. We have used the Black-Scholes option pricing model to estimate grant date fair value for all option grants. The assumptions we use in calculating the fair value of share-based payment awards represent management’s best estimates, but these estimates involve inherent uncertainties and the application of management judgment. As such, as we use different assumptions based on a change in factors, our stock-based compensation expense could be materially different in the future.
Accounting for Income Taxes
We are governed by U.S. income tax laws, which are administered by the Internal Revenue Service (“IRS”). We follow ASC 740, Accounting for Income Taxes, which requires an asset and liability approach to financial accounting and reporting for income taxes. Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases and operating loss and tax credit carryforwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. A valuation allowance is provided when it is more likely than not that some portion or all of a deferred tax asset will not be realized. The ultimate realization of deferred tax assets is dependent upon the generation of future taxable income and the reversal of deferred tax liabilities during the period in which the related temporary difference becomes deductible.
Fair Value of Convertible Notes Payable
We adopted FASB ASU No. 2016-01 “Financial Instruments—Overall (Subtopic 825-10).” In applying ASC 825, it is necessary to determine whether to bifurcate the Beneficial Conversion Feature from the convertible note. Under ASC 825, provided the fixed conversion price stipulated in the convertible note is greater than the fair market value at the date of issuance (“out of the money”), the beneficial conversion feature guidance is not applicable, and the convertible notes are eligible to be valued at fair value and any adjustments recorded in the statement of operations.
We elected to account for the convertible notes payable at fair value with any changes in fair value being recognized in the consolidated statements of operations until the convertible notes are settled. The fair value of the convertible notes is determined with the assistance of a third-party valuation firm. Given the conversion terms that exist, there were two scenarios considered: i) conversion into a preferred share class, or ii) conversion into the common share class. Given the issuance dates, a negotiation discount was calibrated and applied such that the probability weighting of the issued notes is equal to par value as of the respective issuance dates. The probabilities of each conversion scenario were discussed and assigned based on the expectations regarding our future.
Recent Accounting Pronouncements
In December 2019, the FASB issued ASU 2019-12, Income Taxes (Topic 740): Simplifying the Accounting for Income Taxes (ASU 2019-12). ASU 2019-12 removes certain exceptions to the general principles in Topic 740 and also clarifies and amends existing guidance to improve consistency in application. ASU 2019-12 will be effective for public entities for interim and annual periods beginning after December 15, 2020, with early adoption permitted. We adopted ASU 2019-12 and concluded there will be no impact on our consolidated financial statements.
In August 2020, the Financial Accounting Standards Board (FASB) issued Accounting Standards Update (ASU) No. 2020-06, Debt—Debt with Conversion and Other Options (Subtopic 470-20) and Derivatives and Hedging— Contracts in Entity’s Own Equity (Subtopic 815-40): Accounting for Convertible Instruments and Contracts in an Entity’s Own Equity, which simplifies the accounting for convertible instruments by eliminating the requirement to separate embedded conversion features from the host contract when the conversion features are not required to be accounted for as derivatives under Topic 815, Derivatives and Hedging, or that do not result in substantial premiums accounted for as paid-in capital. By removing the separation model, a convertible debt instrument will be reported as a single liability instrument with no separate accounting for embedded conversion features. This new standard also removes certain settlement conditions that are required for contracts to qualify for equity classification and simplifies the diluted earnings per share calculations by requiring that an entity use the if-converted method and that the effect of potential share settlement be included in diluted earnings per share calculations. The new standard will be effective for fiscal years beginning after December 15, 2023 for smaller reporting companies. We have not yet determined the potential impact the adoption may have on our consolidated financial statements.
Off-Balance Sheet Arrangements
We do not engage in transactions that generate relationships with unconsolidated entities or financial partnerships, such as entities often referred to as structured finance or special purpose entities, as a part of our ongoing business. Accordingly, we did not have any off-balance sheet arrangements during any of the periods presented.
Going Concern
Our evaluation of our ability to continue as a going concern requires us to evaluate our future sources and uses of cash sufficient to fund our currently expected operations in conducting research and development activities one year from the date our financial statements are issued. We evaluate the probability associated with each source and use of cash resources in making our going concern determination. The research and development of our diagnostic tests and therapeutic products are inherently subject to uncertainty.
Emerging Growth Company Status
We are both an “emerging growth company” and a “smaller reporting company” as defined by Rule 12b-2 of the Exchange Act and are therefore subject to reduced public company reporting requirements.
As an EGC under the JOBS Act, we may delay the adoption of certain accounting standards until such time as those standards apply to private companies. Other exemptions and reduced reporting requirements under the JOBS Act for EGCs include presentation of only two years of audited financial statements in a registration statement, an exemption from the requirement to provide an auditor’s report on internal controls over financial reporting pursuant to Section 404(b) of the Sarbanes-Oxley Act, an exemption from any requirement that may be adopted by the Public Company Accounting Oversight Board, and less extensive disclosure about our executive compensation arrangements.
In addition, the JOBS Act provides that an EGC can take advantage of an extended transition period for complying with new or revised accounting standards. This provision allows an EGC to delay the adoption of some accounting standards until those standards would otherwise apply to private companies. We have elected to use this extended transition period for complying with new or revised accounting standards that have different effective dates for public and private companies until the earlier of the date we (i) are no longer an emerging growth company or (ii) affirmatively and irrevocably opt out of the extended transition period provided in the JOBS Act. As a result, our financial statements may not be comparable to companies that comply with new or revised accounting pronouncements as of public company effective dates.
We may remain classified as an EGC until the end of the fiscal year following the fifth anniversary of the initial public offering, although if the market value of our Common Stock that is held by non-affiliates exceeds $700 million as of June 30 of any year before that time, or if we have annual gross revenues of $1.235 billion or more in any fiscal year, we would cease to be an EGC as of December 31 of the applicable year. We would also cease to be an EGC if we issue more than $1.0 billion of non-convertible debt over a three-year period.
| 57 |
BUSINESS
Overview
We develop noninvasive diagnostics to detect early-stage lung cancer and other diseases of the lung. We are developing our platform technologies so that, in the future, they could result in broad-spectrum cancer treatments. We develop proprietary noninvasive diagnostic tests using technology that preferentially targets cancer cells and cell populations indicative of a diseased state.
We were formed as a Delaware corporation on March 26, 2014. On June 15, 2016, we formed OncoSelect®, a Delaware limited liability company and our wholly owned subsidiary. On August 14, 2023, we formed PPLS, a Texas limited liability company and our wholly owned subsidiary. Research and optimization of our platform technologies are conducted in our laboratories at The University of Texas at San Antonio.
Our first diagnostic test, CyPath® Lung, addresses the need for noninvasive detection of early-stage lung cancer. Lung cancer is the leading cause of cancer-related deaths. Physicians will be able to order CyPath® Lung to assist in their assessment of patients who are at high risk for lung cancer. The CyPath® Lung test enables physicians to more confidently identify patients who will likely benefit from timely intervention and more invasive follow-up procedures and is another tool to help distinguish them from patients who are likely without lung disease and should continue annual screening. CyPath® Lung has the potential to increase overall diagnostic accuracy of lung cancer, which could lead to increased survival, fewer unnecessary invasive procedures, reduced patient anxiety, and lower medical costs.
Through our wholly owned subsidiary, OncoSelect®, our research has led to discoveries of novel potential cancer therapeutics that specifically and selectively target cancer cells that have been grown in petri dishes.
Recent Developments
On September 18, 2023, PPLS consummated the Acquisition of the Laboratory Assets pursuant to the terms of the Asset Purchase Agreement with Village Oaks and Dr. Joyce. As a result of the Acquisition, the CAP-accredited CLIA-certified clinical pathology laboratory is owned by PPLS. Dr. Joyce was the Medical Director and Laboratory Director of the clinical pathology laboratory prior to the Acquisition and he continues to serve as Medical Director and Laboratory Director after the Acquisition. Founded in 2007 by Dr. Roby Joyce, Village Oaks has provided pathology services to physicians practicing in a variety of outpatient settings. Since September 2021, Village Oaks, under the trade name Precision Pathology Services, has offered CyPath® Lung for sale as an LDT for the detection of early-stage lung cancer. In addition to CyPath® Lung, PPLS intends to continue to offer a range of laboratory services including respiratory testing for SARS-CoV-2 and influenza, anatomical pathology, morphological stains, histological services, DNA extractions, STI testing and women’s and men’s health testing.
Pursuant to the terms of the Asset Purchase Agreement, PPLS acquired all of the Laboratory Assets, which included all of the assets owned by Village Oaks, other than medical assets, which are assets Village Oaks used in connection with its management and operation of a clinical pathology laboratory, now owned by PPLS, and related services business, and assumed certain liabilities and obligations. The laboratory is a clinical pathology laboratory that is CAP-accredited and CLIA-certified. Pursuant to the terms of the Asset Purchase Agreement, Village Oaks received $3,500,000 in consideration for the assets to be purchased by PPLS, of which $1,000,000 was paid by the issuance of 564,972 shares of our restricted Common Stock to the Joyce Trust, which share number was determined by dividing $1,000,000 by $1.77, the average of the trading day closing prices for the thirty (30) days prior to September 15, 2023, rounded to the nearest whole share.
The Asset Purchase Agreement contains customary representations, warranties and covenants made by PPLS and Village Oaks and consummation of the transaction was subject to customary closing conditions, including, among other things, entry into the other ancillary agreements described below.
Pursuant to the Asset Purchase Agreement, PPLS assumed all liabilities and obligations under and obtained any and all rights, title and interest of Village Oaks in and to (i) the Assumed Leases under the Assumption Agreement; (ii) the Assumed Contracts under the Assumption Agreement; (iii) all accounts payable of Village Oaks as of September 18, 2023 that were incurred in the ordinary course of business consistent with past custom and practice; and (iv) the lease of the premises used in connection with operation of the CLIA-certified and CAP-accredited clinical pathology laboratory, pursuant to the Assignment and Assumption of Lease, which Assignment of Lease was consented to by the landlord of the leased premises. The monthly rent is currently $10,143.83 per month and the term of the Lease is five years.
| 58 |
In connection with the Asset Purchase Agreement, PPLS entered into the Management Services Agreement with Village Oaks, pursuant to which PPLS will provide comprehensive management and administrative services to Village Oaks in connection with the operation of Village Oaks’ professional cytopathology, histopathology, clinical and anatomic pathology interpretation medical services practice. PPLS will provide space, equipment, administrative, management and clinical personnel, billing and collection, and related management services to Village Oaks in exchange for a management fee of 70% of the net revenues received by Village Oaks from the provision of the medical services. The Management Services Agreement has an initial term of 20 years and provides that upon expiration of the initial term, it will be automatically extended for two additional successive terms of five years each, unless either party delivers written notice of its intention not to extend the term of the agreement not less than 90 days prior to the expiration of the preceding term. The Management Services Agreement also provides that until the fifth anniversary of its effective date, Village Oaks will not, without the prior written approval of PPLS own, operate or have any financial interest in any other person or entity that operates an independent laboratory or an enterprise within the United States that provides or promotes management or administrative services or any product or services substantially similar to those provided by PPLS.
In connection with the Asset Purchase Agreement, PPLS entered into the Succession Agreement, pursuant to which Dr. Joyce, as holder of 100% of the issued and outstanding stock of Village Oaks, and Village Oaks are restricted from disposing of their equity interests in Village Oaks, subject to certain exceptions, without the prior written consent of us and Village Oaks. The Succession Agreement further provides that the entire equity interest held by Dr. Joyce in Village Oaks will be automatically assigned and transferred to a successor who meets the Eligibility Requirements of a Designated Physician, as such terms are defined and described in the Succession Agreement, in the event of, among other things, the death, disability, retirement, or a court’s determination of incompetence of Dr. Joyce, as well as Dr. Joyce’s failure to satisfy the eligibility requirements of a Designated Physician, exclusion or disqualification from participation in the Medicare program, conviction of a felony or crime or moral turpitude, bankruptcy filing, or material breach of the Succession Agreement. In the event of the automatic transfer of Dr. Joyce’s equity interests in Village Oaks as provided in the Succession Agreement, such agreement provides that the board of directors of Village Oaks shall nominate a group of three candidates as the Designated Physician who satisfy the Eligibility Requirements. In the event the Company desires not to select any of such candidates, the Company shall select and appoint a successor Designated Physician from any other physicians that satisfy the Eligibility Requirements. Subject in all cases to the Management Services Agreement, Dr. Joyce shall not cause any voluntary interruption of the conduct of Village Oaks’ business and operations, and shall use commercially reasonable efforts to preserve (or assist us in preserving) all rights, privileges and franchises held by Village Oaks, including the maintenance of all contracts, copyrights, trademarks, licenses and registrations.
In connection with the Asset Purchase Agreement, PPLS entered into the Professional Services Agreement with Village Oaks, pursuant to which Village Oaks will provide pathology interpretation services as requested on behalf of PPLS based on the professional fees approved for the CPT code for the services provided under the Medicare Physician Fee Schedule in the locality where the test is performed. The Professional Services Agreement has an initial term of 20 years and provides that upon expiration of the initial term, it will be automatically extended for successive terms of 12 months each, unless either party delivers written notice of its intention not to extend the term of the agreement not less than thirty days prior to the expiration of the preceding term.
In connection with the Asset Purchase Agreement, we entered into the Executive Employment Agreement with Dr. Joyce, for a term of three years, to serve as the Medical Director and Laboratory Director of PPLS at a base salary of $333,333.34 per year. Pursuant to the Joyce Employment Agreement, Dr. Joyce was also appointed to serve on our Board of Directors. Dr. Joyce will be eligible to participate in or receive benefits under our benefit plans generally made available to executives of similar status and responsibilities and will be provided use of a company car. In the event the Joyce Employment Agreement is terminated for any reason, including by Dr. Joyce upon 60 days’ notice, by us for cause or by reason of Dr. Joyce’s death, Dr. Joyce (or his estate as applicable) will receive his base salary for the remainder of the three-year employment term. However, the Joyce Employment Agreement provides that if Dr. Joyce breaches any of the restrictive covenants set forth in the Joyce Employment Agreement, including a covenant not to compete during his term of employment and a covenant not to knowingly disclose confidential information, such breach will be grounds for the immediate termination of Dr. Joyce and will result in the forfeiture of all compensation and benefits otherwise due to Dr. Joyce.
One of the Assumed Leases is the Hologic Equipment Lease, pursuant to which PPLS leases reagent equipment from Hologic and is required to purchase a minimum number of specified testing kits each year. The total monthly minimum purchase commitment PPLS is required to pay Hologic, inclusive of the lease of the reagent equipment, is $16,914 per month. The term of the Hologic Equipment Lease currently expires on December 20, 2027.
Another of the Assumed Leases is the Leica Equipment Lease, pursuant to which PPLS leases reagent equipment from Leica and is required to purchase a minimum number of specified testing kits. The total monthly minimum purchase commitment PPLS is required to pay to Leica, inclusive of the lease of the reagent equipment, is $19,790 per month. The term of the Leica Equipment Lease currently expires on March 23, 2026.
| 59 |
One of the Assumed Contracts is the License Agreement. Pursuant to the License Agreement, Pathology Watch granted a license to its digital imaging cloud-based pathology platform to facilitate remote interpretation and billing of pathology specimens by qualified professionals to PPLS for a monthly fee of $25,000. In connection with the License Agreement, Pathology Watch also provides certain support services and marketing vendor services to PPLS for the monthly fee of $38,000, for a total monthly fee paid by PPLS to Precision Watch of $63,000. The License Agreement is for an initial term of 12 months, unless terminated by either party upon 90 days’ notice, and provides that upon expiration of the initial term (or any renewal term), it will be automatically extended for successive 12-month terms, unless either party notifies the other party of its intention not to renew the License Agreement not less than 90 days prior to the expiration of the current term.
In connection with the Asset Purchase Agreement, Dr. Joyce, on behalf of Village Oaks, executed the Bill of Sale, pursuant to which all rights, title, and interest of Village Oaks in and to the permits listed on Exhibit A attached thereto, inclusive of the CLIA-certificate and CAP-accreditation, notwithstanding the transfer of the CLIA certificate by operation of law to PPLS upon consummation of the Acquisition, were confirmed to have been transferred and assigned to PPLS.
Benefits of the Acquisition
Since 2007, Village Oaks, d/b/a Precision Pathology Services, has provided pathology services to physicians practicing in a variety of outpatient settings. Since September 2021, Village Oaks has offered CyPath® Lung for sale as an LDT for the detection of early-stage lung cancer. The Acquisition of the Laboratory Assets supports and enhances our commercial strategy by integrating every aspect of CyPath® Lung, from manufacturing to sales and marketing, and to pathology services and reporting results back to physicians. The Acquisition results in consolidating the royalties to bioAffinity from sale of CyPath® Lung with the revenues derived by PPLS’ sale of the test. The Acquisition provides us with control over essential aspects of the CyPath® Lung test and an opportunity to build scale and efficiency as we expand commercialization of our test. In addition, we anticipate the Acquisition can support our pivotal trial by providing the equipment, experienced laboratory personnel and administrative support, including support for our planned FDA clinical study. Ownership of the Laboratory Assets also provides the clinical services required for us to develop future LDTs that expand our flow cytometry platform, including development of current research into a test for COPD. Village Oaks’ operation of a CLIA-certified and CAP-accredited clinical pathology laboratory has been established over the past 16 years of service. The founder of Village Oaks, Dr. Joyce, will continue as PPLS’ Medical Director and Laboratory Director, thus providing continuity in professional relationships and services. PPLS intends to continue to offer a range of laboratory services in addition to CyPath® Lung, including respiratory testing for SARS-CoV-2 and influenza, anatomical pathology, morphological stains, histological services, DNA extractions, STI testing and women’s and men’s health testing.
Amendment to Warrants
On September 17, 2023, Mr. Girgenti, the Cranye Girgenti Testamentary Trust, Gary Rubin, The Harvey Sandler Revocable Trust, a trust of which Mr. Rubin is a co-trustee, Ms. Zannes and Dr. Joyce consented to an amendment of the terms of the outstanding warrants that they own. Such warrants include warrants (i) Tradeable Warrants to purchase 98,198, 39,182, and 39,182 shares of Common Stock owned by Mr. Girgenti, The Harvey Sandler Revocable Trust, and Ms. Zannes, respectively); (ii) Non-Tradeable Warrants to purchase 102,286, 40,813, and 40,813 shares of Common Stock owned by Mr. Girgenti, The Harvey Sandler Revocable Trust, and Ms. Zannes, respectively; and (iii) Pre-IPO Warrants to purchase 469,063, 8,332, 571,373, 23,571, 17,137, and 14,285 shares of Common Stock owned by Mr. Girgenti, the Cranye Girgenti Testamentary Trust, Mr. Rubin, The Harvey Sandler Revocable Trust, Ms. Zannes and Dr. Joyce, respectively. The Warrant Amendment provides that such warrants will not be exercisable until the date that we file a certificate of amendment to our certificate of incorporation with the State of Delaware which increases the number of shares of our authorized Common Stock to allow for sufficient authorized and unissued shares of Common Stock for the full exercise of all of the outstanding Pre-IPO Warrants, Tradeable Warrants and Non-Tradeable Warrants of the Company and the issuance of all of the shares of Common Stock underlying such warrants.
Financial
To date, we have devoted a substantial portion of our efforts and financial resources to the development of our first diagnostic test, CyPath® Lung. Since our inception in 2014, we have funded our operations principally through public and private sales of our equity or debt securities. On September 6, 2022, we completed our initial public offering of our securities pursuant to which we raised gross proceeds of $7.9 million. As of September 20, 2023, investors participating in the initial public offering exercised a total of 725,576 Tradeable Warrants at a price of $7.35 per share and 310,910 Non-Tradable Warrants at a price of $7.656 per share. Combined with our underwritten public offering, we received an aggregate of approximately $15.6 million as of September 28, 2022. We believe that our available cash together with the proceeds of this Offering will be sufficient to fund our planned operations for at least 12 months following the date of this prospectus.
In the second quarter of 2022, we started to recognize revenue from sales of the CyPath® Lung test by Village Oaks, a CAP-accredited, CLIA-certified clinical pathology laboratory to which we had previously granted a license to develop CyPath® Lung for commercialization and to manufacture, use, market and sell CyPath® Lung as an LDT prior to the Acquisition, which license was assigned to and assumed by PPLS in connection with the Acquisition. We have never been profitable, and as of June 30, 2023, we had total working capital of $7.9 million and an accumulated deficit of approximately $39.9 million. As of June 30, 2023, we had cash and cash equivalents of $8.3 million. We expect to continue to incur significant operating losses for the foreseeable future as we continue the development of our diagnostic tests and therapeutic products and advance our diagnostic tests through clinical trials.
We anticipate raising additional cash needed through the private or public sales of equity or debt securities, collaborative arrangements, or a combination thereof, to continue to fund our operations and develop our products. There is no assurance that any such collaborative arrangement will be entered into or that financing will be available to us when needed in order to allow us to continue our operations, or if available, on terms acceptable to us. If we do not raise sufficient funds in a timely manner, we may be forced to curtail operations, delay our clinical trials, cease operations altogether, or file for bankruptcy.
Organizational Structure
The following organizational chart depicts our principal operating subsidiaries:
| 60 |
Our First Diagnostic Test – CyPath® Lung
Lung cancer remains the most commonly diagnosed cancer and the leading cause of cancer-related deaths worldwide. Globally, there were an estimated 2.1 million lung cancer cases and 1.8 million lung cancer deaths in 2018, as reported by the World Health Organization in its 2018 Cancer Fact Sheet. According to the ALA, screening for individuals at high risk for lung cancer has the potential to improve lung cancer survival rates by finding disease at an earlier stage when it is more likely to be curable. A study published in the New England Journal of Medicine entitled “Survival of patients with stage I lung cancer detected on CT screening” dated October 26, 2006 reported that the survival rate of individuals with Stage I lung cancer who underwent surgical resection within one month after diagnosis had a ten-year survival rate of 92%, as compared to the overall five-year survival rate of 25%. Unfortunately, most lung cancer is detected in late stages. The results of a large national clinical trial that was reported in the New England Journal of Medicine in an article dated August 4, 2011, entitled “Reduced Lung-Cancer Mortality with Low-Dose Computed Tomographic Screening” showed that screening for lung cancer using low-dose computed tomography (“LDCT”) resulted in a reduction of the mortality rate by 20% as compared to screening by x-ray if LDCT screening is used by patients at high risk for lung cancer on an annual basis. Therefore LDCT scans are recommended for screening of an estimated 14 million Americans who are at high risk for lung cancer. If half of these high-risk individuals were screened, over 12,000 lung cancer deaths could be prevented, according to the ALA. However, the New England Journal of Medicine article also reported that LDCT was shown to have a low positive predictive rate of less than 4%. This means that for every 100 people who receive a positive result from LDCT screening and are suspected of having lung cancer, only four of those patients truly have the disease. A reliable, noninvasive and cost-effective diagnostic test can increase diagnosis of early-stage lung cancer while lowering the number of unnecessary and invasive procedures for patients with a false positive result from LDCT screening. (False positive means a person who does not have lung cancer but receives a positive result, in this case from LDCT screening.)
CyPath® Lung is a test for early-stage lung cancer that is designed to meet the need for greater diagnostic certainty. Based on our internal analysis, its use in conjunction with LDCT is predicted to improve the positive predictive value (the probability that patients with a positive LDCT scan truly have the disease) by a factor of five. Our analysis concludes that improving the positive predictive value of LDCT with the use of CyPath® Lung has the potential to subject fewer patients to the stresses of misdiagnosis or unnecessary diagnostic procedures such as biopsies, while also reducing healthcare costs.
CyPath® Lung uses flow cytometry technology to detect and analyze cell populations in a person’s sputum, or phlegm, to find characteristics indicative of lung cancer, including cancer and/or cancer-related cells that have shed from a lung tumor. The flow cytometer is a well-established instrument used in many commercial laboratories that records properties of labeled and unlabeled single cells. Sputum is an excellent sample for analysis because it is in direct contact with any malignancy in the lungs and can thus provide a snapshot of the tumor itself, its microenvironment, and its area of field cancerization. While studies have shown that expert cytological analysis of sputum can detect cancerous and pre-malignant cells, the level of scrutiny required for the analysis is not feasible in the laboratory routine, according to an October 22, 2009 article, “Premalignant and malignant cells in sputum from lung cancer patients,” published in Cancer Cytopathology. The process of looking at microscopy slides is an extremely laborious approach and demands years of expertise. CyPath® Lung uses flow cytometry and automated data analysis developed by AI that allows for an entire sample of sputum to be examined for cost-effective, large-scale screening or diagnosis.
In particular, CyPath® Lung uses a synthetic porphyrin called meso-tetra (4-carboxyphenyl) porphyrin (“TCPP”). Porphyrins are biological pigments that, when exposed to ultraviolet light at certain wavelengths, can result in the cell fluorescing a red or purplish color that can be detected under a microscope or by flow cytometry, according to an article entitled “Laboratory Diagnosis of Porphyria,” published in Diagnostics (Basel) on July 26, 2021. Porphyrins can be man-made, like TCPP, or they can be naturally occurring, like heme that is responsible for the red color in red blood cells. Cancer cells are known to take up certain porphyrins in higher amounts than non-cancer cells, and the high affinity for cancer cells displayed by TCPP makes it an excellent bio-label for cancer, according to an article published in Progress in Clinical and Biological Research in 1984 entitled, “A comparative study of 28 porphyrins and their abilities to localize in mammary mouse carcinoma: uroporphyrin I superior to hematoporphyrin derivative.” As used in CyPath® Lung, the proportion of cells with high TCPP fluorescence intensity in a patient’s sputum sample is a significant predictor of lung cancer. We hold multiple patents protecting our use of TCPP for the diagnosis, monitoring, and treatment of cancer. In addition, we have multiple domestic and foreign patent applications to protect the use of flow cytometry and our AI-developed automated analysis platform in the detection of lung cancer and other lung diseases using sputum as a sample.
We developed an algorithm as part of a test validation trial that used machine learning to distinguish samples from high-risk patients who had lung cancer from those who are cancer-free. Village Oaks developed CyPath® Lung for sale as an LDT in accordance with the standards of the CAP and the regulations and guidance of the CLIA program, which is administered by CMS. Our test can analyze an average sputum sample containing about 14 million cells in approximately 20 minutes using integrated software for high-throughput, user-friendly standardized analysis of flow cytometric sample data. A physician’s report is generated within minutes after data acquisition. The test can be put into routine lab use without requiring expert evaluation of samples or being subject to operator bias. Our approach allows the entire sputum sample to be rapidly analyzed. The numerical analysis developed with machine learning captures complex interactions between lung cancer, the microenvironment, and areas of field cancerization that would be difficult if not impossible for individuals to predict or detect reliably by eye. For example, during test development, we discovered that viability staining density suggests a link with apoptosis, or cell death, that is linked to many cancers, including lung cancer. Our model also suggests that specific markers of immune cell populations may be informative as to the presence of cancer in the lung. These findings are the result of our machine learning approach to automated analysis.
The CyPath® Lung diagnostic process uses sputum that is obtained noninvasively in the privacy of a patient’s home. Physicians can order the test for patients they suspect have lung cancer or patients with a positive LDCT screening result. A patient collects his or her sample using a hand-held, noninvasive assist device, ICU Medical’s acapella® Choice Blue, that acts to break up mucus in the lungs and help a person cough up sputum from the lung into a collection cup. The acapella® Choice Blue has been 510(k) cleared by the FDA as a positive expiratory pressure device to help mobilize lung secretions in people with certain lung conditions. The sputum sample is shipped overnight to a clinical pathology laboratory that is accredited by the CAP and certified under the CLIA program, and processed with CyPath® Lung that includes antibodies that distinguish different cell types and the synthetic porphyrin TCPP that identifies cancer cells and/or cancer-associated cells. Proprietary automated analysis software developed by bioAffinity Technologies analyzes sample data in minutes, resulting in a patient report provided to the physician who orders the test. CyPath® Lung can be used by physicians to find early-stage lung cancer in their patients who have undergone lung cancer screening.
We conducted a 150-patient test validation trial of people at high risk for lung cancer including patients with the disease (N=28) and those cancer-free (N=122) that resulted in CyPath® Lung’s overall 88% specificity, meaning the ability to correctly identify a person without cancer, and 82% sensitivity, meaning the ability to correctly identify cancer in a person with the disease. For the subset of patients in this trial who had lung nodules smaller than 20mm or no nodules at all, this trial resulted in 92% sensitivity and 87% specificity. In this subset of 132 individuals with small nodules, 119 patients were cancer-free and 13 had confirmed lung cancer. The detection of small lung nodules in people who have early-stage cancer can increase lung cancer survival.
| 61 |
In this 19-month test validation trial participants provided a sputum sample and were released from the study after a physician either confirmed the individual was cancer-free by examination of CT imaging or confirmed the presence of lung cancer by biopsy. Flow cytometry and patient data used in the analysis produced results that included (1) the proportion of cells with a high ratio of high TCPP fluorescence intensity over cell size; (2) the proportion of cells with an intermediate ratio of fluorescence intensity caused by the viability dye (FVS510) over cell size; (3) the proportion of cells that were CD206 negative but positive for one or more of the following markers: CD66b (granulocytes), CD3 (T cells), and CD19 (B cells); and (4) patient age.
The CyPath® technology is based on research originating at Los Alamos National Laboratory in collaboration with St. Mary’s Hospital (Colorado) in which cancer samples were differentiated from non-cancer samples with 100% accuracy. This early research was conducted with sputum from 12 uranium miners. Microscope slides were made of the sputum samples labeled with the active ingredient of CyPath®, the synthetic and fluorescent porphyrin TCPP. The Los Alamos research study of 12 uranium miners included eight men with cancer and four healthy individuals. Researchers were blinded to the sample origin and looked for the presence of highly fluorescent cells indicating uptake of TCPP and the presence of lung cancer. The length of the study and specific follow-up was not reported, but researchers did report that one patient entering the study as a healthy subject was correctly diagnosed with cancer by the test.
We conducted market research with pulmonologists, oncologists, cardiothoracic surgeons, radiologists, and internists engaged in the diagnosis and treatment of lung cancer to help assess these stakeholders’ reactions to the new diagnostic test. Research revealed a strong interest in CyPath® Lung, driven by the high level of unmet clinical need for noninvasive diagnostics. A survey conducted with 240 pulmonologists and internists, the primary audience for the test, showed that 96% would use CyPath® Lung if it were available today as an adjunct to LDCT screening and diagnosis. Physicians responded favorably to a noninvasive diagnostic technology that gives them more confidence in their decision to proceed with more aggressive follow-up procedures if the test comes back positive. If test results are negative, physicians could rule out lung cancer, thus reducing the number of costly invasive procedures that result from the LDCT false-positive rate.
Physicians can order the CyPath® Lung laboratory test for use by people at high risk for lung cancer who are recommended for annual screening by LDCT. While LDCT is shown to lower the mortality rate of lung cancer by at least 20% as compared to x-ray screening, the LDCT screening method has a low positive predictive value that can result in many people undergoing unnecessary invasive diagnostic procedures to confirm or rule out the presence of lung cancer. A physician who orders a CyPath® Lung test can have greater confidence in determining the next steps in patient care. Noninvasive sample collection and the test’s three-day turnaround in providing patient results after sample receipt make CyPath® Lung well suited for both sophisticated and less developed markets. On June 6, 2023, the AMA approved a Current Procedural Terminology (“CPT”) Proprietary Laboratory Analysis (“PLA”) code specifically for use with CyPath® Lung, which code was publicly released on June 30, 2023. The new CPT code will be effective October 1, 2023. Prior to and in the interim until the new code is effective, CyPath® Lung is reported with a non-specific CPT code, for which payment is determined by the payer on a case-by-case basis. Payment for the new PLA code was discussed on July 19, 2023, by the Medicare Advisory Panel on Clinical Diagnostic Laboratory Tests. CMS’ preliminary determination will be released in September 2023 followed by a 30-day comment period. A 2024 payment determination, effective January 1, 2024, will be made by CMS in November 2023. There is an opportunity for reconsideration in next year’s payment cycle.
We have an agreement with GO2 Partners to produce patient collection kits and to provide warehousing and distribution services for sending out the kits. Laboratory reagents, supplies and equipment are commercially available through multiple vendors. Sample processing, labeling, and data collection can be accomplished by a laboratory technician skilled in general laboratory techniques. Data analysis leading to a physician’s report is done by automated analysis software fully integrated into the test.
To our knowledge, CyPath® Lung is the first cancer diagnostic that combines automated flow cytometric analysis to predict the presence of lung cancer from sputum samples.
OncoSelect® Therapeutics Research
OncoSelect® Therapeutics, LLC, a Delaware limited liability company and our wholly owned subsidiary, is a preclinical-stage biopharmaceutical discovery company with a focus on therapeutics that deliver cytotoxic (cell-killing) effects on a broad selection of human cancers from diverse tissues while having little or no effect on normal cells.
Unlike many of our industry competitors, OncoSelect® does not pursue therapies that depend on specific mutations, biomarkers, or other genetic or epigenetic abnormalities for their effect. We pursue research based on our own scientific discoveries demonstrating that inhibition of the expression of two specific cell membrane proteins results in the selective killing of various cancer cell types grown in the laboratory with little or no effect on normal (non-cancerous) cells.
Our scientific discoveries stemmed from research we conducted to better understand the mechanism by which TCPP, the synthetic porphyrin used in CyPath® Lung, selectively enters cancer cells. We have established several specific areas of therapeutic research that have evolved from our TCPP experiments.
OncoSelect® therapies offer the possibility of broad applications in cancer treatment. OncoSelect® will use a licensing business model for selective chemotherapeutic compounds to be developed by the Company.
Clinical Validation, Certification, and Classification of CyPath® Lung
A 19-month test validation clinical trial of CyPath® Lung collected sputum noninvasively from 150 people at high risk for lung cancer, including patients with the disease (N=28) and those cancer-free (N=122). Patients collected their sputum sample over three days at home before bringing their sample to the clinical collection site. Samples were shipped overnight to the laboratory for analysis. Study participants in the high-risk cohort had a CT to confirm they did not have lung cancer. Those in the cancer cohort had imaging and a biopsy that confirmed lung cancer. After providing a sputum sample, participants were released from the study after a physician either confirmed the individual was cancer-free by examination of CT imaging or confirmed the presence of lung cancer by biopsy. Flow cytometry and patient data used in analysis to produce the results included (1) the proportion of cells with a high ratio of high TCPP fluorescence intensity over cell size; (2) the proportion of cells with an intermediate ratio of fluorescence intensity caused by the viability dye (FVS510) over cell size; (3) the proportion of cells that were CD206 negative but positive for one or more of the following markers: CD66b (granulocytes), CD3 (T cells), and CD19 (B cells); and (4) patient age.
| 62 |
As reported in an article entitled “Detection of Early-Stage Lung Cancer in Sputum using Automated Flow Cytometry and Machine Learning,” published in Respiratory Research on January 21, 2023, the overall analysis resulted in 88% specificity and 82% sensitivity. More than half of those in the cancer cohort had lung cancer in the earlier Stages I-II. The analysis, performed on an LSRII flow cytometer, resulted in 92% sensitivity and 87% specificity in the subgroup of these patients (N=132) who had no nodules or lung nodules smaller than 20 mm on their LDCT scan, while eight out of 10 (80%) of Stage I tumors were correctly identified. Sensitivity is the percentage of persons with the disease – in this case lung cancer – who are correctly identified by the test. Specificity is the percentage of persons without lung cancer who are correctly identified by the test. The cancer group included all lung cancer types, but mostly squamous cell carcinoma and adenocarcinoma lung cancer (in near equal numbers), showing that CyPath® Lung detects all types of lung cancer.
Following completion of the test validation trial, CyPath® Lung was evaluated independently by Village Oaks, which developed the test for sale as an LDT in accordance with CAP/CLIA standards. An LDT is a type of IVD test that is developed, validated, and performed within a single laboratory. CyPath® Lung has been validated and, until consummation of the Acquisition, has been developed, commercialized and marketed by Village Oaks, a CAP-accredited, CLIA-certified clinical pathology laboratory in San Antonio, Texas, pursuant to a joint development agreement with us. In third quarter 2022, Village Oaks was inspected by CAP in accordance with CAP/CLIA regulatory standards and regulations resulting in continued accreditation for the laboratory and the CyPath® Lung test as an LDT.
As part of CAP/CLIA certification, Village Oaks evaluated the performance of CyPath® Lung employing its own laboratory technicians and a different flow cytometer, the Navios EX. Results of Village Oaks’ certification were comparable to those from the test validation trial and demonstrated that CyPath® Lung remains robust to differences in sample handling, processing, and the type of flow cytometer.
We intend to voluntarily seek FDA clearance of the CyPath® Lung as a Class II IVD medical device for the detection of lung cancer which will enable us to market our CyPath® Lung directly to U.S, physicians and their patients and facilitate dialogues with payors. We have designed our pivotal trial with guidance from our CRO, Courante Oncology, and we are preparing a pre-submission that will be submitted to the FDA for review and feedback. We anticipate a three-year diagnostic trial including an 18-month patient enrollment of approximately 1,800 patients. Similar to the test validation trial, the planned pivotal trial will analyze flow cytometry and patient data including (1) the proportion of cells with a high ratio of high TCPP fluorescence intensity over cell size; (2) the proportion of cells with an intermediate ratio of fluorescence intensity caused by the viability dye (FVS510) over cell size; (3) the proportion of cells that were CD206 negative but positive for one or more of the following markers: CD66b (granulocytes), CD3 (T cells), and CD19 (B cells); and (4) patient age. Patient enrollment is scheduled to begin in early 2024 at up to 20 collection sites. Assuming the study is successful, we intend to submit a de novo classification request to the FDA within six months of study completion.
The Patient- and Physician-Friendly CyPath® Lung Process
CyPath® Lung is designed to be noninvasive and patient friendly. The diagnostic process uses sputum that is obtained noninvasively in the privacy of a patient’s home. We anticipate that physicians will most often order the test for their patients after lung cancer screening reveals a lung nodule considered to be indeterminate because of the nodule size and lack of suspicious characteristics. Indeterminate lung nodules are noncalcified, 7 to 20 mm in diameter, and with a cancer risk between 5% and 60%, according to an article published in Cancer Prevention Research on December 1, 2014, entitled “Indeterminate Pulmonary Nodules: Risk for Having or for Developing Lung Cancer?”
For the CyPath® Lung test, patients are given a small sample collection kit during an office visit with their physician. (See Figure 1 below.) From the privacy of the patient’s home, the patient collects a sample of his or her sputum over three days using a hand-held assist device that comes in the collection kit called an acapella® Choice Blue made by ICU Medical. The acapella® acts to break up mucus in the patient’s lung and facilitates the patient’s ability to cough up sputum from the lung into a collection cup that is also supplied with the kit. In addition to the kit’s step-by-step instructions, an instructional video and a live patient coach are available by calling 855-MYLUNGS to help patients with sample collection. After the three-day sample of sputum is collected in the collection cup, the patient puts the collection cup into the kit and uses a pre-addressed envelope provided in the kit to overnight the sample to the laboratory.
At the laboratory, the sputum is processed by technicians into a single-cell suspension and labeled with TCPP, which preferentially binds to cancer cells and/or cancer-related cells. Cells are also stained with fluorescently labeled antibodies that identify hematopoietic and epithelial cells within the sputum sample. A viability dye is used to eliminate dead cells from analysis. The sputum sample is run through a flow cytometer that collects data on individual cells and cell populations. An average sputum sample containing about 14 million cells can be profiled in approximately 20 minutes. A laboratory technician skilled in general laboratory techniques can accomplish sample processing, labeling, and data collection. Automated analysis software produces a physician report in minutes that is provided to the attending physician, The report stratifies the patient into one of two risk groups. Those patients deemed “likely or very likely” to have cancer may benefit from aggressive intervention. Those “unlikely or very unlikely” to have a malignancy may continue imaging surveillance. The physician also receives a numerical score between 0.1 to 1.0, with 0.1-0.5 being a negative result and 0.6 to 1.0 considered positive for lung cancer.
Physicians receive test results within three days after the laboratory receives the patient’s sputum sample. CyPath® Lung testing helps identify patients who should undergo more aggressive follow-up procedures to confirm a suspected lung cancer. When CyPath® Lung sample analysis determines a patient is unlikely or very unlikely to have lung cancer, the result can serve to support a physician’s decision to monitor this patient by following a recommended LDCT screening routine.
| 63 |
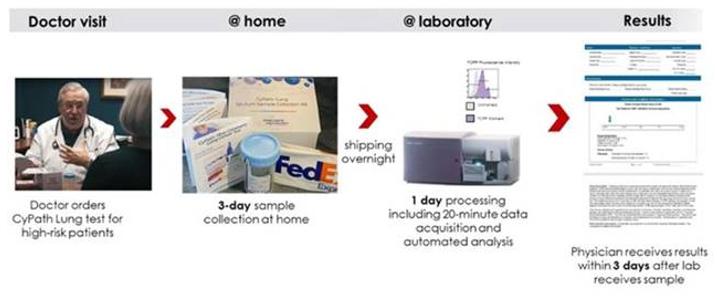
Figure 1. Patient- and Physician-Friendly CyPath® Lung Process.
CyPath® Lung Research and Clinical Studies
The high affinity of TCPP for cancer and cancer-related cells and its fluorescent nature makes it an excellent bio-label for cancer. The CyPath® Lung technology is based on this concept and scientific work originating at Los Alamos National Laboratory in collaboration with St. Mary’s Hospital in Colorado. In the Los Alamos research study, sputum samples from lung cancer patients were differentiated from non-cancer samples with 100% accuracy. This early research was conducted with sputum from 12 uranium miners. Microscope slides of sputum samples were labeled with the synthetic and fluorescent porphyrin TCPP. The Los Alamos research study of 12 uranium miners included eight men with cancer and four healthy individuals. Researchers were blinded to the sample origin and looked for the presence of highly fluorescent cells indicating uptake of TCPP as an indicator of lung cancer. The length of the study and specific follow-up was not reported, but researchers did report that one patient entering the study as a healthy subject was correctly diagnosed with cancer by the test. Later, a blinded clinical trial was conducted and results published September, 2015, in an article entitled “Early Detection of Lung Cancer with Meso-Tetra (4-Carboxyphenyl) Porphyrin-Labeled Sputum” in the Journal of Thoracic Oncology. This study reported on an earlier version of CyPath® Lung that used a fluorescent microscope to directly identify cells labeled with TCPP in one-third or less of the sputum sample. For each trial participant, researchers manually scanned 12 microscope slides labeled with TCPP for the presence of red fluorescent cells (“RFCs”) displaying a spectral signature that indicated uptake of TCPP in the cell. In addition to measuring the spectral signature, the fluorescent intensity and cell size of RFCs were measured. The test data, including fluorescent intensity over cell size, was analyzed. The trial was conducted over 24 months and resulted in 81% test accuracy, 77.9% sensitivity, and 65.7% specificity in the ability to correctly differentiate between samples from lung cancer patients and those at high risk who were cancer-free. The Patriquin trial required participants to provide a sputum sample and CT imaging of the lungs. Those in the cancer cohort underwent a biopsy to confirm lung cancer. High-risk patients displaying indeterminate nodules were followed for 18 months to confirm they were cancer-free. The Patriquin study concluded that optimizing the test to provide for analysis of the entire sputum sample would improve results.
We continued development of our lung cancer diagnostic technology culminating in a flow cytometry-based CyPath® Lung test incorporating automated AI analysis that evaluates the entire sputum sample. A blinded diagnostic trial of the advanced test resulted in 92% sensitivity and 87% specificity in the subgroup of these patients (N=132) who had no nodules or lung nodules smaller than 20 mm on their LDCT scan, while eight out of 10 (80%) of Stage I tumors were correctly identified. The cancer group included all lung cancer types, but mostly squamous cell carcinoma (N=13) and adenocarcinoma (N=11) lung cancer, showing that CyPath® Lung detected all types of lung cancer.
All CyPath® Lung studies and clinical trials studies performed to date are summarized in the table below.
CyPath® Lung Studies and Clinical Trials
| Study Description | Results | |
| Porphyrin’s localization and evaluation of cancer cell uptake of four different porphyrins | TCPP porphyrin localizes more than other porphyrins in cancer cells; higher uptake of TCPP in cancer cells than in normal cells. Uptake was determined by visual assessment. Cell lines were used. Researchers did not report the length of time taken to conduct this study, nor any follow-up. | |
| Blinded study to diagnose lung cancer by labeling sputum with TCPP and identifying red fluorescing cells under a microscope | Study of uranium miners (cancer N=8 / healthy N=4) that labeled sputum with TCPP resulted in 100% sensitivity and 100% specificity. Classification of cancer was made by subjective visual assessment of the presence and intensity of red fluorescing cells on slides. In this blinded study, one patient initially enrolled as a healthy subject was correctly diagnosed with cancer by the test. Length of study not reported. No patient follow-up was reported except for correct detection of cancer in patient initially enrolled as healthy. |
| 64 |
| Internal validation study with microscopy-based assay completed to optimize TCPP labeling of sputum containing cancer and cancer-related cells in lung cancer samples | In this research study lasting eight months, the florescence intensity of TCPP-labeled cells in sputum was measured by subjective visual assessment of microscope slides to distinguish samples from cancer and healthy cohorts. Researchers who were blinded to sample origin correctly identified samples from lung cancer patients (cancer N=15 / healthy N=12) resulting in 100% sensitivity and 100% specificity. Participants were not followed up after providing the sputum sample and CT scan or biopsy. |
| Early Detection of Lung Cancer with Meso-Tetra (4-Carboxyphenyl) Porphyrin-Labeled Sputum, published in Journal of Thoracic Oncology in September 2015 | A 24-month clinical trial of 128 high-risk smokers and cancer patients used microscopy-based assay to identify TCPP-labeled cells in sputum (cancer N=26 / high risk N=102) that resulted in 81% accuracy, 77.9% sensitivity, 65.7% specificity. Slides were scanned. Fluorescent intensity and cell size of RFCs were objectively measured by software. High-risk participants who were cancer-free were followed for 18 months to confirm status. | |
| Sputum analysis by flow cytometry; an effective platform to analyze the lung environment, published in PLOS One on August 17, 2022 | Research reporting on the CyPath® Lung test’s quality controls included manual analysis of cell population data acquired by flow cytometry analysis of sputum. This research evaluated flow cytometry data from 164 participants’ sputum samples analyzed manually for differences in cell characteristics, cell population size, and cell fluorescence intensity. | |
|
Detection of Early-Stage Lung Cancer in Sputum using Automated Flow Cytometry and Machine Learning, published in Respiratory Research on January 21, 2023
|
Test validation clinical trial using bioAffinity’s automated flow cytometry CyPath® Lung test (cancer N=28 / high risk N=122) resulted in overall 82% sensitivity and 88% specificity for the test; CyPath® Lung sensitivity is 92% and specificity is 87% for patients with lung nodules smaller than 20 mm.
|
|
| Porphyrin-modified beads for use as compensation controls in flow cytometry, published in Journal of Visualized Experiments JoVE on March 24, 2023 | Reporting on the protocol for preparing porphyrin-labeled compensation beads invented by bioAffinity and used to optimize the results of CyPath® Lung test to detect early-stage lung cancer. |
The Cancer Diagnostics Market and CyPath® Lung
The global cancer diagnostic market is projected to grow from an estimated $102.24 billion in 2022 to $162.57 billion in 2030, with a compound annual growth rate (“CAGR”) of 6.1%, according to a market research report issued by Research and Markets in June 2023. A January 2023 report, also by Research and Markets, stated that the market worldwide for lung cancer diagnostic tests was estimated at $2.6 billion in 2022 and is projected to reach $4.7 billion by 2030, with a CAGR of 7.8% over 2022-2030. We have the potential to play a significant role in the cancer diagnostic market because our platform is noninvasive, cost-effective, and has a potential to lead to better patient outcomes.
CyPath® Lung is a commercial diagnostic tool for detecting early-stage lung cancer. In 2018, we licensed CyPath® Lung to Village Oaks, which began marketing CyPath® Lung in Texas in April 2022 as an LDT in accordance with CAP/CLIA regulations pursuant to the terms of the joint development agreement between us and Village Oaks. Limited funds were available to market CyPath® Lung until we completed our initial public offering in September 2022. CyPath® Lung is sold to physicians who order CyPath® Lung for patients at high risk for lung cancer after an LDCT confirms the presence of lung nodule(s).
On June 6, 2023, the AMA approved a Current Procedural Terminology (CPT) Proprietary Laboratory Analysis (PLA) code specifically for use with CyPath® Lung, which code was publicly released on June 30, 2023. The new code will be effective October 1, 2023. Prior to and in the interim until the new code is effective, CyPath® Lung is reported with a non-specific CPT code, for which payment is determined by the payer on a case-by-case basis. Payment for the new PLA code was discussed on July 19, 2023, by the Medicare Advisory Panel on Clinical Diagnostic Laboratory Tests. CMS’ preliminary determination will be released in September 2023 followed by a 30-day comment period. A 2024 payment determination, effective January 1, 2024, will be made by CMS in November 2023. There is an opportunity for reconsideration in next year’s payment cycle.
As a front-end diagnostic tool used in conjunction with LDCT, our lung cancer test is designed to help determine whether more expensive, specialized, and/or invasive tests are warranted. CyPath® Lung compares favorably to current standards of care for diagnosing lung cancer, including invasive biopsies, as seen in the table shown below.
| 65 |
Comparison of CyPath® Lung to Current Standards of Care
| Diagnostic
Test or Procedure |
Intended Patient | Sensitivity | Specificity | Procedural Risk | Source | |||||||||
| CyPath® Lung | High risk | 82 | % | 88 | % | None | “Detection of Early-Stage Lung Cancer in Sputum using Automated Flow Cytometry and Machine Learning,” published in Respiratory Research on January 21, 2023 | |||||||
| CyPath® Lung | High risk – nodules less than 20 mm | 92 | % | 87 | % | None | “Detection of Early-Stage Lung Cancer in Sputum using Automated Flow Cytometry and Machine Learning,” published in Respiratory Research on January 21, 2023 | |||||||
| Low Dose CT screening | High risk | 93.80 | % | 73.40 | % | Radiation exposure | “Results of initial low dose computed tomographic screening for lung cancer,” published in the New England Journal of Medicine on May 23, 2013 | |||||||
| FDG PET imaging | Suspicious lung nodules | 89 | % | 75 | % | Radiation exposure | “Accuracy of FDG-PET to diagnose lung cancer in areas with infectious lung disease: a meta-analysis,” published in JAMA in September 2014 | |||||||
| Bronchoscopy | Suspicious lung nodules – central lesions | 88 | % | 47 | % | Invasive, risk of collapsed/bleeding lung infection |
“A bronchial genomic classifier for the diagnostic evaluation of lung cancer,” published in the New England Journal of Medicine on July 16, 2015 | |||||||
| Fine Needle Biopsy | Suspicious lung nodules | 90.4 | % | 75.4 | % | Invasive, risk of collapsed/bleeding lung infection |
“Fine-needle aspiration biopsy versus core-needle biopsy in diagnosing lung cancer: a systemic review,” published in Current Oncology in February 2012 | |||||||
| Core Needle Biopsy21 | Suspicious lung nodules | 89.1 | % | 88.6 | % | Invasive, risk of collapsed/bleeding lung infection |
“Global patterns and trends in lung cancer incidence: a population-based study,” published in the Journal of Thoracic Oncology on February 16, 2021 | |||||||
Our business model is to immediately address the need for a quick-to-market, noninvasive, cost-effective lung cancer diagnostic that will save lives and reduce medical costs. The U.S. Preventive Services Task Force recommended new guidelines for screening in March, 2021, nearly doubling the number of Americans at high risk for lung cancer who are recommended for annual screening to 14 million people, according to the ALA. China has an estimated 300 million smokers according to the World Health Organization. In Europe, it is estimated that there is one new case of lung cancer diagnosed every minute, with incidence rates for males the highest in East-European countries and a five-year survival rate of only 13%, as reported by a May 2021 article, “Lung cancer screening in Europe: where are we in 2021?,” published in Translational Lung Cancer Research. We expect to pursue CE marking of CyPath® Lung for sale in the European Union.
We conducted market research with pulmonologists, oncologists, cardiothoracic surgeons, radiologists, and internists engaged in the diagnosis and treatment of lung cancer to help assess these stakeholders’ reactions to the new diagnostic, CyPath® Lung. Research revealed a strong interest in CyPath® Lung, driven by the high level of unmet clinical need for noninvasive diagnostics. A survey conducted with 240 pulmonologists and internists, the primary audience for the test, showed that 96% would use CyPath® Lung if it were available today as an adjunct used for diagnosis after LDCT screening. Physicians see the value of a noninvasive diagnostic technology with the ability to confirm or rule out cancer and reduce the number of costly invasive procedures that result from LDCT’s low positive predictive rate.
Joint Development Agreement with Village Oaks
We entered into a joint development and project agreement (the “Joint Development Agreement”) with Village Oaks in October 2018 to develop CyPath® Lung as an LDT under CLIA. In connection with the Acquisition, Village Oaks assigned all right, title and interest in the Joint Development Agreement to PPLS. The Joint Development Agreement contains details concerning each party’s duties and responsibilities in the development project. Under the agreement, we are solely responsible for: (i) conducting all research and clinical trials; (ii) conducting all sales and marketing activities; (iii) preparing and implementing all advertising; (iv) attending all medical and industry conferences; (v) training all personnel; and (vi) procuring, assembling, labeling and storing the product. PPLS is solely responsible for: (i) all steps necessary to validate CyPath® Lung for purposes of CLIA; (ii) receiving and handling all CyPath® Lung test kits returned for processing; (iii) sending out all bills and claims in connection with the sale and processing of the CyPath® Lung test and evaluating the results; (iv) communicating test results to physicians; and (v) properly equipping, maintaining and staffing laboratories to process the CyPath® Lung tests. We are also required to refer inquiries from prospective customers seeking to purchase CyPath® Lung to PPLS and to use commercially reasonable efforts to recommend such prospective customers purchase all of their requirements from PPLS.
The Joint Development Agreement contains a license by us to PPLS of our intellectual property associated with CyPath® Lung to allow PPLS to manufacture, use, market and sell the CyPath® Lung LDT only in those U.S. states and territories where PPLS is permitted under applicable law to do so. Prior to the Acquisition, the license required Village Oaks to pay us a royalty of 50% of the gross revenue received by Village Oaks from the sale and processing of the CyPath® Lung test; however the consummation of the Acquisition will convert the royalties to an intercompany transaction that will be eliminated upon consolidating our financial statements with those of PPLS. The Joint Development Agreement also provides that we will be the sole owner of any intellectual property that is developed by either party during the joint development project, regardless of inventorship. We are solely responsible for preparing, filing, prosecuting and maintaining all patent applications and patents covering any jointly developed intellectual property.
The Joint Development Agreement remains in effect until we obtain FDA approval to directly commercialize CyPath® Lung or its functional equivalent, unless terminated earlier by either party in connection with the other party’s breach of the agreement, insolvency or bankruptcy. All licenses granted under the Joint Development Agreement terminate upon the termination of the Joint Development Agreement. If we obtain FDA approval (or its functional equivalent) to directly commercialize CyPath® Lung, we have agreed to appoint PPLS as a distributor of the FDA-approved test pursuant to the terms of a mutually acceptable distribution agreement.
| 66 |
CyPath® Lung Business Development Plan
We believe in the viability of our Business Plan based on the circumstances surrounding our business that are known to us as of the date of this prospectus. However, the timing, strategies, and stages of our Business Plan may evolve in light of new circumstances that cannot be predicted with certainty at this time. Our Business Plan envisions four phases of expanding market entry into the U.S., the EU, and worldwide that are timed to maximize our resources and minimize market risk. Phase 1 of our Business Plan has already begun with a limited market launch of our LDT CyPath® Lung in south Texas. This limited test market launch is designed to evaluate our marketing program and help us ensure each step in the care pathway – from the initial order by physicians to sputum collection and processing, to generating and delivering the patient report – is efficient and effective. This limited test market approach allows us to refine future positioning and develop strategic insight for our CyPath® Lung test before expanding to a larger market. The next step in our marketing plan provides for and will be followed by expansion into the Southwest market area in 2024 followed by a staged nationwide expansion of sales and marketing. In addition to introducing pulmonologists, family practitioners, and other providers to CyPath® Lung, we are selling CyPath® Lung tests to the Department of Defense (DOD) to conduct an observational study, “Detection of Abnormal Respiratory Cell Populations in Lung Cancer Screening Patients Using the CyPath® Lung Assay,” and for research and development on using bronchoalveolar lavage fluid as a biological sample to assess cardiopulmonary function and exercise performance in military personnel post COVID-19 infection. The DOD represents a significant potential market for CyPath® Lung. In February 2023, we announced that the marketing and advertising firms of Havas Health & You and Trinity Life Sciences had been engaged to build the CyPath® Lung brand and position it for success in the cancer diagnostics sector. Havas Health & You, the world’s largest global health network, is creating the branding and broader marketing strategy to align with the need for a patient-friendly diagnostic that gives physicians another tool to assess the potential or presence of lung cancer in their high-risk patients. Trinity Life Sciences is providing advisory services, insights, and analytics to our marketing strategy for CyPath® Lung. Phase 2 of our Business Plan anticipates entering the EU market with CyPath® Lung as a CE-marked IVD test with sales in the Netherlands, followed by a staged EU expansion. Phase 3 of our Business Plan focuses on the marketing of an FDA-cleared CyPath® Lung test, beginning with a pivotal clinical trial in the U.S. Phase 4 of our Business Plan accelerates the market presence of CyPath® Lung in countries in Asia, Eastern Europe, and Australia after obtaining FDA marketing authorization.
At each phase of commercialization, we plan to develop messaging and marketing programs, including key convention attendance, digital marketing, social media presence, and advertising, to create an “inbound” lead generation mechanism that delivers our message to our target audience. In addition, we plan to collaborate with key opinion leaders (“KOLs”) to expand our third-party reference and speaking pool of experts. We will provide support and collateral materials, including posters, presentations, videos, and peer-reviewed papers, to our KOLs who will present data and their experience with CyPath® Lung at key meetings. This content can be shared across platforms, including websites and sales tools, and will be used as references to support our product claims as well as sales and marketing efforts to physicians, reference laboratories, and patients. We will also work with lung cancer advocacy groups throughout all phases to support the message that routine screening can diagnose cancer at an early stage and save lives.
The Competition for CyPath® Lung
In 2022, we evaluated 67 companies advancing tests for the early detection of lung cancer that provided at least a scientific foundation for their tests. These competitors are investigating lung cancer screening and diagnostic methods that use various types of collected samples (blood, breath, nasal epithelial cells, saliva, sputum, and urine) or imaging systems. Of those 67 companies, we found that only 11 had conducted clinical studies in a manner and with results that could lead to further analysis. The majority of these 11 tests are in research and development, with only four tests on the market and one available to a limited number of medical centers. Although CyPath® Lung was never tested directly against any of these five tests, comparison of the published performance numbers suggests CyPath® Lung might outperform them all. Furthermore, CyPath® Lung is noninvasive—not even requiring a needle stick—and cost-efficient, and processing and analysis procedures are easy to perform. The 11 tests are discussed below in more detail.
Based on published data and results of clinical trials, we grouped lung cancer diagnostic tests into three categories: 1) balanced tests; 2) rule-out tests, and 3) rule-in tests. Balanced tests aim at excluding patients without cancer from unnecessary follow-up diagnostic procedures and detecting patients with early-stage cancer who can proceed to more aggressive procedures to confirm diagnosis. Balanced tests can be the most cost effective. Those that perform well, like CyPath® Lung, are most useful to a physician and his or her patient because they provide the most information, allowing a quicker decision on what follow-up path to choose, i.e., whether to move forward with more aggressive follow-up procedures (i.e., when the CyPath® Lung test reveals a “likely” or “highly likely” cancer result) or to stay more conservative (i.e., when the CyPath® Lung test reveals an “unlikely” or “very unlikely” cancer result). Rule-out tests aim to exclude patients without cancer from unnecessary follow-up procedures with high accuracy (if the test provides a “negative” result), but among the remainder of patients who do not receive an unambiguous negative result, there is still uncertainty about who has cancer and who does not. Cancer patients for whom time is of the essence are included in this group of patients still in uncertainty. The patient can lose precious time with a rule-out test. Rule-in tests aim to identify patients with cancer but in doing so may identify many people without cancer as positive. Therefore, rule-in tests have a low positive predictive value. Rule-in and rule-out tests are less useful as well-performing balanced tests.
From the 67 companies we evaluated, we found only seven tests, including CyPath® Lung, that represent a balanced test for early lung cancer detection and that have advanced to the point that there is sufficient data for evaluation. Of our six competitors with well-balanced tests (two sell the same test; one in the U.S. and one in China), four companies (20/20 GeneSystems; Nuclexi; Savicell; Visongate) conducted their studies on a population that does not match the high-risk population for which the test is intended. Their clinical data, therefore, is not necessarily representative of the results that would be achieved in the population of patients who actually will use the test.
The two remaining balanced tests are not on the market. One of these latter tests Is LungLB, a FISH-based test that requires a significant amount of experience to conduct. LungLife AI (in the U.S.) and SanMed Biotech (in China) offer the LungLB test (in China, it is called the MDA Test). The test uses a visualization instrument from BioView to read microscope slides. Studies from both companies have been consistent in result, but with significantly lower performance than a study from the original inventor, perhaps an indication of the difference in expertise between our laboratory and that of the inventor. LungLB is developing an automated system of analysis, but additional clinical trials are necessary to determine the efficacy of the test. The second balanced test is EPN Scan, developed by IONIQ Sciences, formerly known as proLung Dx. This test requires unique, expensive equipment. The clinical trials that tested the performance of the EPN scan have provided inconsistent results. An early trial with 41 patients showed promise, but later clinical trial results were considerably less impressive.
There was insufficient data reported for the EPN Scan to determine the Area Under the Curve (the “AUC”), a key indicator of a test’s ability to discriminate between cancer and non-cancer. In general, an AUC of 0.5 suggests no ability to distinguish between people with cancer and people without cancer. An AUC of 0.7 to 0.8 is considered acceptable, 0.8 to 0.9 is considered excellent, and more than 0.9 is considered outstanding. CyPath® Lung trials have resulted in AUC of 0.89 and 0.9. The two trials conducted for the LungLB/MDA Test for which there is data resulted in an AUC of 0.82
Two rule-out tests are currently on the market while one is available to a limited number of medical centers. Both the REVEAL, offered by MagArray, and Nodify-XL2, offered by Biodesix, are rule-out tests, meaning the tests aims to exclude patients without cancer. The REVEAL test is a blood test intended for patients with indeterminant nodules. In their 97-patient clinical validation trial, only patients with an intermediate risk of cancer, based either on a physician’s judgement or a clinical model, took part. This requirement led to 30% of high -risk patients being excluded at the onset of their analysis. In addition, the positive predictive value of the REVEAL test was 13.5% as compared to CyPath® Lung’s positive predictive value of 43.2%. Importantly, no patients were excluded from the CyPath® Lung test. The tests had negative predictive values of 98% and 97.8%, respectively. The second rule-out test, Nodify-XL2, is used only by people with a pre-test probability of cancer less than 50%. As with the REVEAL test, a large number of patients were excluded from analysis. In the case of Nodify-XL2, about 55% of patients with lung nodules that physicians considered indeterminate, namely lung nodules sized between 8-30 mm, were excluded from the study. In addition, Nodify XL-2 reported an AUC of 0.62 (unacceptable) and 0.76 (acceptable) for their two clinical trials, as compared to CyPath® Lung with an AUC of 0.89 and 0.90 in two independent study groups (excellent). Finally, the Percepta nasal swab test offered by Veracyte is currently available to a limited number of medical centers but expected to be fully launched in 2022. Initial performance parameters for this test were developed on samples obtained from people scheduled to undergo bronchoscopy. In this case, the AUC was not provided. The positive predictive value was only 16.9% and the negative predictive value was 99.3%.
| 67 |
The only rule-in test on the market is EarlyCDT Lung that has not reported the AUC for its two clinical trials. EarlyCDT Lung reports a positive predictive value of 34.5% as compared to CyPath® Lung’s 43.2% positive predictive value. In addition, there is a still a 10% chance for a person with a negative EarlyCDT test to have cancer. Thus, neither a positive nor negative EarlyCDT test result provides much more certainty after a positive LDCT screening.
We believe there are many reasons why CyPath® Lung is a superior test when compared to its competitors. First, lung sputum is an excellent medium for early lung cancer detection because sputum is in close contact with the tumor and pre-cancerous areas that shed cancer and pre-cancerous cells directly into the sputum, can be obtained noninvasively, and can be transported easily. Moreover, sputum contains immune cell populations in reaction to the presence of a tumor. Second, ours proprietary technology is straightforward. Our CyPath® Lung platform technology is not a molecular test and does not collect genetic material that requires immediate processing. CyPath® Lung uses well-established flow cytometry techniques to investigate cells contained in the sputum for characteristics that indicate whether cancer is present. Sample processing is straightforward, and laboratory technicians can be easily trained. Reagents used by the test are widely available. Data acquisition and analysis is fully automated, allowing for efficient test results. Third, CyPath® Lung has shown high specificity and sensitivity that is similar to far more invasive and more expensive procedures currently used to detect lung cancer. Fourth, CyPath® Lung is cost effective. Existing CPT cost codes that have a reimbursable track record have been identified for use with CyPath. Fifth and as important as any of our test’s benefits, CyPath® Lung is patient friendly, providing at-home sample collection that is noninvasive and offers particular benefit during a public healthcare crisis like the coronavirus pandemic.
Research and Development Activities
We are continuing our research and development activities pertaining to diagnostics that include multiple studies we believe will support FDA final approval of CyPath® Lung, which we will seek after the pivotal trial is complete. We are also conducting research toward the development of CyPath® Lung for detection of COPD and the assays use with bronchoalveolar lavage fluid (BAL), with support from the DOD. With regard to therapeutic research, we continue our experiments focused on establishing proof-of-concept for our discovery that the silencing or knockdown of two genes that each encode a cell surface receptor results in cancer death without much harm to healthy cells.
Other Diagnostic Applications for the CyPath® Platform
We expect to expand our platform technology to detect and monitor other lung diseases.
Chronic Obstructive Pulmonary Disease and Other Diseases of the Lung. The respiratory diagnostics market was valued at $8.2 billion in 2021 and is expected to reach $13.8 billion by 2030, according to a market research study published by Custom Market Insights in November 2022. The World Health Organization reports that COPD is the third leading cause of death in the world, causing nearly 3.23 million deaths in 2019. The disease is characterized as an abnormal inflammatory response and airflow obstruction that cannot be fully reversed. Early detection allows for the use of therapies when the disease is less severe, which slow the progression of the disease. We plan to build on our expertise in using sputum as a sample for flow cytometric analysis to develop a test to detect COPD at an early stage and monitor for signs of impending exacerbations before clinical signs occur. CyPath® Lung’s flow cytometry platform provides for identification of cell populations and other parameters of disease in the lung. Our test illuminates the microenvironment of the lung. We believe that our flow cytometric test can be designed to identify other lung diseases, such as COPD and asthma, using antibodies that characterize cell populations in sputum specific to the disease.
OncoSelect® Therapeutic Platforms
We plan to pursue our therapeutics business through our wholly-owned subsidiary, OncoSelect® that is a preclinical-stage biopharmaceutical discovery company with a focus on therapeutics that deliver cytotoxic (cell-killing) effects on a broad selection of human cancers from diverse tissues while having little or no effect on normal cells.
The Cancer Therapeutic Market
It is undeniable that cancer is a very complex disease. The Centers for Disease and Control Prevention reports that cancer was the second leading cause of death, after heart disease in the United States in 2020, with 602,350 cancer deaths recorded. While improvements in early cancer diagnosis have had an impact on five-year survival rates in some cancers, the prognosis for patients with advanced or metastatic disease remains poor.
According to the EvaluatePharma World Preview 2020, Outlook to 2026 report, the worldwide market for oncology drugs has shown steady growth in recent years and is projected to continue at a CAGR of 20.2% between 2019 and 2026. Oncology drug revenue is the highest of all pharmaceutical indications, with projected oncology drug sales projected to reach a value of $394.24 billion by 2027, up from $141.33 billion in 2019, as reported by Fortune Business Insights in its Oncology Drugs Market Research Report, 2020-2027. In June 2023, MarketWatch reported that the global market for RNA therapeutics, which include antisense and RNA interference drugs such as siRNAs, is projected to grow from $1.33 billion in 2021 to $1.94 billion in 2027 at a CAGR of 6.43%.
Our discoveries have opened new opportunities to develop various drug combination therapies targeting multiple cancer vulnerabilities simultaneously. We believe we are in a unique position to take advantage of this growing market.
Drugs targeting specific genetic aberrations in cancer cells have been widely pursued, but their efficacy is often limited by the development of drug resistance due to genetic or epigenetic changes or their applicability to select patient populations. As an alternative to the drug targeting of genetic aberrations, some researchers have begun to refocus on underlying factors that are common to many cancers, such as the altered cancer metabolism in cancer cells. At OncoSelect®, we are not pursuing therapies that are dependent upon specific gene mutations or other genetic and epigenetic abnormalities for their effect. We are pursuing research based on our own scientific discoveries.
| 68 |
From Diagnostic Research Comes A Key Therapeutic Discovery
Our therapeutic platforms originated from our research on how TCPP, the porphyrin used in CyPath® Lung, enters cancer cells. We conducted research to better understand the mechanism of TCPP’s selective uptake in cancer cells. Our research identified cell-membrane proteins which capture small molecules outside of the cell and bring them inside the cell, called receptors, that are associated with TCPP, Experiments that we conducted confirmed that at least two of these receptors called CD320 and LRP2, contributed to TCPP uptake by cancer cells. When these receptors were individually “knocked down” in cancer cells and therefore could not be made by the cell, TCPP uptake was significantly decreased. Knock-down of CD320 and LRP2 receptors was achieved by introducing siRNA molecules into the cells that cause the destruction of CD320 and LRP2 gene products. These gene products were the messenger (m)RNAs that are the precursors of the receptor protein. An siRNA is a small, chemically synthesized piece of RNA that specifically binds to mRNA, prohibiting the further production of the corresponding proteins. Thus, the reduction of CD320 or LRP2 mRNAs reduced the CD320 or LRP2 protein, respectively, and resulted in decreased TCPP uptake in a variety of cancer cells, with a larger decrease observed when CD320 was knocked down. We subsequently discovered that the simultaneous knockdown of these two cell-surface receptors, CD320 and LRP2, was deadly to cancer cells or inhibited their growth significantly but left normal cells virtually unharmed.
siRNAs can be easily synthesized and are easily introduced into cells growing in a petri dish by a process called transfection. siRNAs have been broadly adopted by academic and industrial researchers for the fundamental study of the function of genes and their proteins. We designed siRNAs to effectively eliminate CD320 and LRP2 protein production to study their role in TCPP uptake into the cell. With these CD320 and LRP2 siRNAs, we achieved a reduction of CD320 and LRP2 protein levels of up to 90%. Simultaneous siRNA knock-down of CD320 and LRP2 in normal cells, including skin fibroblasts and breast epithelial cells, did not affect cell growth. However, knock-down of CD320 and LRP2 in cancer cell lines derived from diverse tissues (lung, breast, prostate, brain, and skin cancers) inhibited cell growth or killed the cells, in some cases up to 80%. The Figure below compares cells that were left alone (no treatment in the upper row pictures to cells treated with CD320/LRP2 siRNA treatment. Interestingly, in some cell lines, when either CD320 or LRP2 were silenced individually, a concurrent increase in protein expression of the other receptor was observed, suggesting that CD320 and LRP2 compensate for each other’s function; hence, silencing both receptors is required for optimal cell killing. These discoveries can lead to novel and very promising therapeutic approaches for diverse cancers that do not appear to be dependent on any aberrant genetic or epigenetic profiles.
bioAffinity Technologies’ Therapeutic Discovery:
Killing Cancer Cells with Little or No Harm to Normal Cells
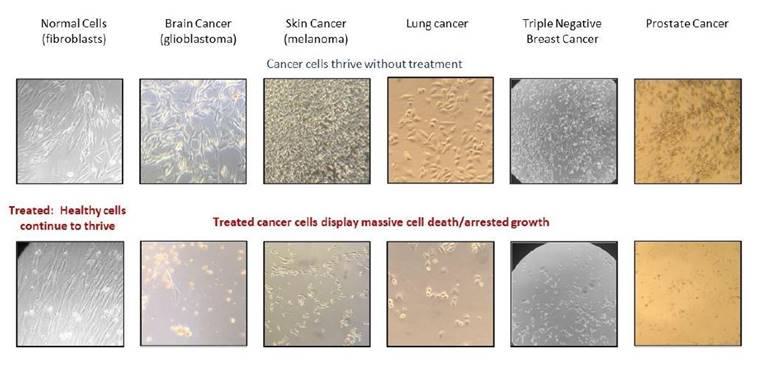
Research at OncoSelect® continues the optimization of the siRNAs used in knocking down the CD320 and LRP2 receptors and testing their performance in additional cell lines grown in the laboratory. As reported in a July 2022 article published in Pharmaceutics, entitled “Nanoparticles-Based Strategies to Improve the Delivery of Therapeutic Small Interfering RNA in Precision Oncology,” recent strategies have also been undertaken related to improving siRNA delivery for therapeutic efficacy. We seek to develop this technology to the advanced preclinical-stage and undertake further development in conjunction with a partner who has greater clinical trial capabilities and expertise with siRNA delivery systems. Our ultimate goal is the development of a new class of cancer therapeutics with broad applicability in diverse human cancers.
Industry Opportunity
The global market for cancer diagnostic tests is expected to grow dramatically in coming years. According to a 2021 Global Cancer Diagnostics Market Research Report, cancer diagnostic tests, including devices, grew from $156.27 billion in 2020 to $170.21 billion in 2021, with a compound annual growth rate of 8.9%, and is projected to reach $239.23 billion in 2025. Lung cancer is the most common cancer globally and its incidence continues to increase in some large nations including China, where lung cancer is the leading cause of cancer-related morbidity and mortality, as reported in the Journal of Thoracic Oncology in October 2020 in an article entitled “Lung Cancer in People’s Republic of China.” According to a 2023 report “Lung Cancer Diagnostics: Global Strategic Business Report,” the global market for lung cancer diagnostic tests was estimated at $2.6 billion in 2022 and is projected to reach a value of $4.7 billion by 2030, with a compounded annual growth rate of 7.8% over 2022-2030. Clinical diagnostics play an important role in disease prevention, detection, and management. Our first test, CyPath® Lung, focuses on the leading cause of cancer death among both men and women. Lung cancer accounted for approximately 18% of all cancer deaths worldwide in 2020, as reported by the World Health Organization. Lung cancer typically may not be symptomatic in its early stages when it is most treatable. An estimated 14 million patients at high risk for lung cancer in the U.S. are recommended for annual screening. Initially, physicians would order CyPath® Lung for those high-risk patients as an adjunct to LDCT screening to aid in the decision whether to pursue more aggressive follow-up procedures. A more accurate and reliable lung diagnostic pathway using LDCT and noninvasive methods could result in fewer patients being subjected to the stresses of unnecessary, invasive diagnostic procedures such as biopsies. CyPath® Lung is well suited for use in both sophisticated and less-developed markets because sample collection is noninvasive and conducted at home, the sample can be shipped overnight by commercial carriers and sample processing and automated analysis can be completed by laboratory technicians skilled in general laboratory techniques. Patient reports are provided to the ordering physician within three days of sample receipt at the laboratory.
| 69 |
The Competition for Our Therapeutic Products
Interest in the RNAi therapeutics space has fluctuated since discovery of the phenomenon in the 1980s. This is likely due to the challenge of delivering siRNA drugs in the body and directing them to the tumor cells. There are several reasons for this: the chemical composition of siRNAs do not allow them to easily cross into cells from the outside. Second, proteins in the fluid component of blood (plasma) can damage and degrade the siRNAs, destroying them before they reach their intended destination. A third difficulty is that siRNAs have problems traveling to their target after they get into cells, and only a very small fraction of siRNAs that enter cells actually arrive at their intended destination within the cell to perform their therapeutic function. However, in the last seven years, the patent activity and capital investment in this area have increased considerably, especially after the approval of the first RNAi therapeutic, ONPATTRO (developed by Alnylam Pharmaceuticals), in August 2018 by the FDA and European Medicines Agency.
Current competitors in the application of the RNAi strategy against cancer include Arrowhead Pharmaceuticals, which is investigating preclinically an siRNA/dynamic polyconjugate (DPC) peptide combination, ARC-HIF2, targeting Hif-2α for renal cell carcinoma. Arbutus Biopharma is investigating a proprietary lipid nanoparticle technology for siRNA delivery, leading to a potential siRNA therapeutic (TKM-PLK1) targeted toward hepatocellular carcinoma and possibly other cancers. TKM-PLK1 is in multiple Phase I/II trials at this time. Dicerna may have a candidate in an expanded Phase I clinical trial for solid tumors (DCR-MYC). Silence Pharmaceuticals, Silenseed, and Apeiron Biologics separately report RNAi-based oncology drugs in Phase IIA clinical trials for pancreatic cancer.
The potential of the double knockdown strategy is exemplified by the July 6, 2021 announcement from Sirnaomics, Inc., describing FDA approval to launch a Phase I clinical trial for their lead product candidate, STP707, an anti-cancer siRNA therapeutic, in subjects with advanced/metastatic or refractory solid tumors. STP707 takes advantage of a dual-targeted inhibitory property in which simultaneously knocking down TGF-β1 and COX-2 gene expression in the tumor microenvironment increases active T cell infiltration. As in our studies with CD320 and LRP2 knockdown, each individual siRNA was demonstrated to inhibit the expression of their target mRNAs, and combining the two siRNAs produced a synergistic effect (which, in the case of STP707, was shown to diminish pro-inflammatory factors). Over-expressions of TGF-β1 and COX-2 have been well-characterized in playing key regulatory roles in tumorigenesis. The company also employed a proprietary polypeptide nanoparticle (PNP)-enhanced targeted delivery to solid tumors and metastatic tumors via systemic administration. With respect to the market viability of therapeutics in this class, we note that, to date, four siRNA-based therapies have been approved by the FDA (patisiran, givosiran, lumasiran and inclisiran) and six other candidates are in clinical trials, according to a January 2023 article entitled “Small Interfering RNA (siRNA)-Based Therapy.
Competitive Strengths
We conduct an ongoing competitive analysis of companies in the lung cancer diagnostic sector of the clinical diagnostics market. In 2022, we evaluated companies that reported an interest in diagnosing lung cancer, focusing on 67 companies and academic institutions we identified as active in the early lung cancer diagnostic sector. A thorough evaluation of the early lung cancer diagnostic landscape reveals multiple reasons why CyPath® Lung is positioned to be a market leader. CyPath® Lung performance shown in our test validation trial resulted in 92% sensitivity and 87% specificity in high-risk patients who had lung nodules 20 mm or smaller. Eight out of 10 (80%) Stage I tumors were correctly identified, indicating that CyPath® Lung can find lung cancer at its earliest stage. Overall, when diagnosing lung cancer in all stages, the clinical trial resulted in CyPath® Lung specificity of 88% and sensitivity of 82%, similar to far more invasive procedures and surgery currently used to diagnose lung cancer. (See the “Comparison of CyPath® Lung to Current Standards of Care” chart in the “Business” section of this prospectus.) The test validation trial of 150 patients was conducted over 19 months. Participants provided a sputum sample and were released from the study after a physician either confirmed the individual was cancer-free by examination of CT imaging or confirmed the presence of lung cancer by biopsy. Test data used to produce results included: (1) the proportion of cells with a high ratio of high TCPP fluorescence intensity over cell size; (2) the proportion of cells with an intermediate ratio of fluorescence intensity caused by the viability dye (FVS510) over cell size; (3) the proportion of cells that were CD206 negative but positive for one or more of the following markers: CD66b (granulocytes), CD3 (T cells), and CD19 (B cells); and (4) patient age.
The majority of competitors’ tests either incorrectly classify a high proportion of people without cancer as having the disease (known as false negatives) more than 50% of the time or misdiagnose people as cancer-free (known as false positives) more than 50% of the time. It is important to note that most competitors who have conducted clinical trials also have not designed their trials to evaluate the test’s measure of accuracy – such as sensitivity and specificity – in the high-risk population for whom the test is intended A patient collects his or her sample at home, which is a particular benefit during a pandemic. Sample processing for CyPath® Lung can be done by laboratory technicians, and reagents used by the test are widely available. Data acquisition and analysis and test results are fully automated.
| 70 |
Corporate Information
We were incorporated in the State of Delaware on March 26, 2014. Our principal executive office is located at 22211 West Interstate 10, Suite 1206, San Antonio, Texas 78257, and our telephone number at that address is (210) 698-5334. Our website address is https://www.bioaffinitytech.com/. Information contained on or that can be accessed through our website is not incorporated by reference into this prospectus. Investors should not consider any such information to be part of this prospectus.
Intellectual Property Portfolio
We strive to protect the proprietary technologies that we believe are important to our business, including pursuing and maintaining patent protection intended to cover our commercialized diagnostic test, pipeline product candidates and their use, as well as other inventions that are important to our business. In addition to patent protection, we also protect valuable company assets with copyright, trademark, trade secret, and know-how through confidentiality agreements, invention assignment agreements, and a trade secret program to protect aspects of our business that are not amenable to, or that we do not consider appropriate for, patent protection. The confidentiality agreements are designed to protect our proprietary information, and the invention assignment agreements are designed to gain company control and ownership of technologies that are developed for us by our employees, consultants, or other third parties. We seek to preserve the integrity and confidentiality of our data and trade secrets by maintaining physical security of our premises, physical and electronic security of our information technology systems, and non-disclosure agreements with those that produce or receive company confidential information. While we have confidence in our agreements and security measures, either may be breached, and we may not have adequate remedies. In addition, our trade secrets may otherwise become known or independently discovered by competitors.
Our commercial success depends in part upon our ability to obtain and maintain patent and other proprietary protection for commercially important technologies, inventions, and trade secrets related to our business, defend and enforce our intellectual property rights, particularly our patent rights, preserve the confidentiality of our trade secrets, and operate without infringing valid and enforceable intellectual property rights of others.
The patent positions for biotechnology companies like us are generally uncertain and can involve complex legal, scientific, and factual issues. In addition, the coverage claimed in a patent application can be significantly reduced before a patent is issued, and its scope can be reinterpreted and even challenged after issuance. As a result, we cannot guarantee that any of our product candidates will be protectable or remain protected by enforceable patents. We cannot predict whether the patent applications we are currently pursuing will issue as patents in any particular jurisdiction or whether the claims of any issued patents will provide sufficient proprietary protection from competitors. Any patents that we hold may be challenged, circumvented, or invalidated by third parties.
As of September 1, 2023, we and OncoSelect® have a patent estate that includes 15 issued U.S. and foreign counterpart patents including two U.S. patents and thirteen foreign counterpart patents in Australia, Canada, China, France, Germany, Hong Kong, Italy, Mexico, Spain, Sweden, and the United Kingdom. We and OncoSelect® own all patents and trademarks in its intellectual property portfolio. One U.S. patent and nine counterpart foreign patents directed at diagnostic applications expire in 2030. Therapeutic patents have been registered in Australia China, Hong Kong, Mexico and the U.S. that expire in 2037.
With regard to our diagnostic test called CyPath® Lung and other diagnostic test candidates, we have one issued U.S. patent and nine foreign counterpart patents in Canada, China, France, Germany, Hong Kong, Italy, Spain, Sweden, and the United Kingdom. With regard to our diagnostic patent applications, there are two families of which one is directed at diagnosing lung health using flow cytometry and the other is directed at proprietary compensation beads used in analysis by flow cytometry. The diagnostic patent family of pending applications that is directed at diagnosing lung health includes one pending non-provisional U.S. patent application and eight foreign counterpart patent applications in Australia, Canada, China, European Patent Office, Japan, Hong Kong, Mexico, and Singapore filed in 2019, one International Patent Application filed in 2022 and one International Patent Application filed in 2023. Also, a patent application directed at the composition of compensation beads was filed as an International Patent application in 2022.
With regard to our therapeutic product candidates, we have one issued U.S. patent, four issued foreign patents, two pending U.S. applications, and nine foreign applications pending in Canada, China, European Patent Office, Hong Kong, India, and Japan and one pending International Patent Application filed in 2022. The therapeutic intellectual property is made up of two families, including one family directed at our siRNA product candidates for the treatment of cancer, and another family directed at our porphyrin conjugates for treating cancer. One therapeutic patent application has been granted in China that expires in 2037.
The term of individual patents depends upon the legal term of the patents in the countries in which they are obtained. In most countries in which we file, the patent term is 20 years from the earliest date of filing a non-provisional patent application. In the U.S., the term of a patent covering an FDA-approved drug may be eligible for a patent term extension under the Hatch-Waxman Act as compensation for the loss of patent term during the FDA regulatory review process. The period of extension may be up to five years beyond the expiration of the patent, but cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval. Only one patent among those eligible for an extension may be extended, and a given patent may only be extended once. Similar provisions are available in Europe and in certain other jurisdictions to extend the term of a patent that covers an approved drug. It is possible that issued U.S. patents covering each of our therapeutic product candidates may be entitled to patent term extensions. If our product candidates receive FDA approval, we intend to apply for patent term extensions, if available, to extend the term of patents that cover the approved product candidates. We also intend to seek patent term extensions in any jurisdictions where they are available; however, there is no guarantee that the applicable authorities, including the FDA, will agree with our assessment of whether such extensions should be granted, and, if granted, the length of such extensions.
| 71 |
In addition to patent protection, we also rely on know-how and trade secret protection for our proprietary information that is not amenable to, or that we do not consider appropriate for, patent protection, to develop and maintain our proprietary position. However, trade secrets can be difficult to protect. Although we take steps to protect our proprietary information, including restricting access to our premises and our confidential information, as well as entering into agreements with our employees, consultants, advisors, and potential collaborators, third parties may independently develop the same or similar proprietary information or may otherwise gain access to our proprietary information. As a result, we may be unable to meaningfully protect our know-how, trade secrets, and other proprietary information.
In addition, we plan to rely on regulatory protection based on orphan drug exclusivities, data exclusivities, and market exclusivities.
Government Regulation
United States
Diagnostic Products (including medical devices and tests)
In the United States, medical devices, including in vitro diagnostic products (“IVDs”) are subject to extensive regulation by the FDA, under the FDCA and its implementing regulations, and certain other federal and state statutes and regulations. The laws and regulations govern, among other things, the design, manufacture, storage, recordkeeping, approval, labeling, promotion, post-approval monitoring and reporting, distribution and import and export of medical devices, including IVDs. IVDs are a category of medical device that can be purchased by clinical laboratories and used to perform laboratory testing. IVDs include reagents and instruments used to detect the presence of certain chemicals or other biomarkers in human specimens for the purpose of diagnosis or detection of diseases or conditions. IVDs can also be used to perform predictive, prognostic, and screening testing. Like other medical devices, IVDs may require premarket review and clearance, authorization or approval by the FDA. Failure to comply with applicable requirements may subject a device and/or its manufacturer to a variety of administrative and judicial sanctions, such as FDA refusal to approve pending PMA applications, issuance of warning letters or untitled letters, mandatory product recalls, import detentions, civil monetary penalties, and/or judicial sanctions, such as product seizures, injunctions, and criminal prosecution.
Laboratory-Developed Tests
CyPath® Lung has entered the U.S. market as an LDT. The FDA considers LDTs to be tests that are developed, validated and performed within a single laboratory. The FDA has historically taken the position that it has the authority to regulate LDTs as IVDs under the FDCA, but it has generally exercised enforcement discretion with regard to LDTs. This means that even though the FDA believes it can impose IVD regulatory requirements on LDTs, such as requirements to obtain premarket approval, authorization, or clearance, it has to date generally chosen not to enforce those requirements. Although the FDA has generally exercised enforcement discretion for LDTs, the FDA has asserted authority over LDTs when the FDA deems it appropriate to address significant public health concerns. Separately, CMS oversees clinical laboratory operations through the CLIA program.
FDA has indicated that it may seek to increase its regulation of LDTs through rulemaking. The Spring 2023 Agenda of Regulatory and Deregulatory Actions, published semi-annually by the Office of Management and Budget (OMB), indicates that FDA plans to issue a Notice of Proposed Rulemaking in the Federal Register in August 2023 “to make explicit that laboratory developed tests (LDTs) are devices under the Federal Food, Drug, and Cosmetic Act,” although it is unclear whether this will occur.
Legislative proposals addressing the FDA’s oversight of LDTs have been previously introduced. In March 2020, the VALID Act was introduced in the Senate, which proposes a regulatory framework for IVDs and LDTs and would require premarket approval for some in vitro clinical tests, including LDTs that are not currently reviewed by the FDA. The VALID Act was reintroduced in July 2021. In March 2020, the VITAL Act was introduced in the Senate, which would expressly shift the regulation of LDTs from the FDA to CMS. The VITAL Act was reintroduced in May 2021. VALID was again reintroduced in March 2023. Neither VALID nor VITAL has been enacted. The likelihood that Congress will pass the VALID Act, VITAL Act, or similar legislation, and the extent to which such legislation may affect the FDA’s plans to regulate LDTs as medical devices, by either giving the FDA explicit authority to do so or, alternatively, stating that the FDA does not have authority to regulate LDTs, is difficult to predict. It is possible that the FDA’s general policy of enforcement discretion for LDTs may be changed in the future, such that FDA clearance or approval of LDTs is required.
Clinical Laboratory Improvement Amendments of 1988
Clinical laboratories testing specimens collected in the U.S. for the purpose of disease diagnosis or health assessment are subject to CLIA, unless exempt. CLIA establishes quality standards for all clinical laboratory testing to ensure the accuracy, reliability and timeliness of patient test results regardless of where the test was performed. In particular, these regulations mandate that clinical laboratories must be certified by the federal government or an accreditation organization with deemed status from the federal government, or must be located in a state that has been granted exemption from CLIA requirements because the state has in effect laws that provide for requirements equal to or more stringent than CLIA requirements. CLIA also requires that laboratories meet quality assurance, quality control and personnel standards, perform proficiency testing and undergo inspections. The CLIA standards applicable to clinical laboratories are based on the complexity of the testing performed by the laboratory, which ranges from “waived” to “moderate complexity” to “high complexity.” In the case of tests performed using IVDs, test complexity categorization of the IVD is performed by the FDA.
CAP is a member-based physician organization comprising approximately 18,000 board-certified pathologists. CAP’s Laboratory Accreditation Program has been granted deeming authority from the federal government, meaning that CAP accreditation can be used to qualify for CLIA certification and to satisfy CLIA inspection requirements.
Medical Devices
The FDCA classifies medical devices into one of three categories based on the risks associated with the device and the level of control necessary to provide reasonable assurance of safety and effectiveness. Class I devices are low risk and are subject only to general regulatory controls. Class II devices are moderate risk. They are subject to general controls and may also be subject to special controls. Class III devices are generally the highest risk devices. They are required to obtain premarket approval and comply with postmarket conditions of approval in addition to general regulatory controls.
| 72 |
Generally, establishments that design and/or manufacture devices are required to register their establishments with the FDA. They also must provide the FDA with a list of the devices that they design and/or manufacture at their facilities.
The FDA enforces its requirements by market surveillance and periodic inspections, both announced and unannounced, to review records, equipment, facilities, laboratories and processes to confirm regulatory compliance. These inspections may include the manufacturing facilities of subcontractors. Following an inspection, the FDA may issue a report, known as a Form 483 notice of observations, listing instances where the manufacturer has failed to comply with applicable regulations and/or procedures. The FDA may also issue a public warning letter. If the manufacturer does not adequately respond to a Form 483 or warning letter, the FDA may take enforcement action against the manufacturer or impose other sanctions or consequences, which may include:
| ● | cease and desist orders; | |
| ● | injunctions, or consent decrees; | |
| ● | civil monetary penalties; | |
| ● | recall, detention or seizure of products; | |
| ● | operating restrictions, partial or total shutdown of production facilities; | |
| ● | refusal of or delay in granting requests for 510(k) clearance, de novo classification, or premarket approval of new products or modified products; | |
| ● | withdrawing 510(k) clearances, de novo classifications, or premarket approvals that are already granted; | |
| ● | refusal to grant export approval or export certificates for devices; and | |
| ● | criminal prosecution. |
Premarket Authorization and Notification
While most Class I and some Class II devices may be marketed without prior FDA authorization, many Class II and most Class III medical devices can be legally sold within the U.S. only if the FDA has: (i) approved a PMA application prior to marketing, generally applicable to most Class III devices; (ii) cleared the device in response to a premarket notification (a “510(k) submission”), generally applicable to some Class I and most II devices; or (iii) authorized the device to be marketed through the de novo classification process, generally applicable for novel low or moderate risk devices. PMA applications, 510(k) premarket notifications, and de novo requests require payment of user fees.
510(k) Premarket Notification
Product marketing in the U.S. for most Class II and a limited number of Class I devices typically follows the 510(k) premarket notification pathway. To obtain 510(k) clearance, a manufacturer must submit a premarket notification demonstrating that the proposed device is substantially equivalent to a legally marketed device, referred to as the “predicate device.” A predicate device may be a previously 510(k) cleared device or a Class III device that was in commercial distribution before May 28, 1976 for which the FDA has not yet called for PMA applications, or a product previously placed in Class II or Class I through the de novo classification process. The manufacturer must show that the proposed device has the same intended use as the predicate device, and that it either has the same technological characteristics, or has different technological characteristics but is shown to be equally safe and effective and does not raise different questions of safety and effectiveness as compared to the predicate device.
The FDA has a user fee goal to apply no more than 90 calendar review days to 510(k) submissions. During the process, the FDA may issue an Additional Information request, which stops the clock. The applicant has 180 days to respond, although during the COVID-19 Public Health Emergency, the FDA has permitted companies an additional 180 days in which to respond. Therefore, the total review time absent the Public Health Emergency could be up to 270 days, and in practice may be longer.
After a device receives 510(k) clearance, any modification that could significantly affect its safety or effectiveness, or that would constitute a major change in its intended use, requires a new 510(k) clearance or could require a PMA approval or de novo classification. The FDA requires each manufacturer to make this determination in the first instance, but the FDA can review any such decision. If the FDA disagrees with a manufacturer’s decision not to seek a new 510(k) clearance for the modified device, the agency may retroactively require the manufacturer to seek 510(k) clearance, de novo classification, or PMA approval. The FDA also can require the manufacturer to cease marketing and/or recall the modified device until 510(k) clearance or PMA approval is obtained.
De Novo Classification
Devices of a new type that the FDA has not previously classified based on risk are automatically classified into Class III regardless of the level of risk they pose. To avoid requiring PMA review of novel low- to moderate-risk devices classified in Class III by operation of law, Congress enacted a provision that allows the FDA to reclassify a novel low- to moderate-risk device into Class I or II in the absence of a predicate device that would support 510(k) clearance. The FDA evaluates the safety and effectiveness of devices submitted for review under this de novo pathway and devices determined to be Class II can serve as predicate devices for future 510(k) applicants. The de novo pathway can require clinical data.
The FDA has a user fee goal to review a de novo request in 150 calendar review days. During the process, the FDA may issue an Additional Information request, which stops the clock. The applicant has 180 days to respond. Therefore, the total review time could be as long as 330 days, and in practice may be longer. During the COVID-19 Public Health Emergency, applicants have been given an additional 180 days in which to respond.
| 73 |
PMA Approval
A Class III product generally must follow the PMA approval pathway. The PMA must be supported by sufficient valid scientific evidence, including clinical study data, to assure that the device is safe and effective for its intended use(s). After completion of clinical testing, a PMA including the results of all non-clinical, clinical, and other testing and information relating to the product’s marketing history, design, labeling, manufacture, and controls, is prepared and submitted to the FDA.
The PMA approval process is generally more expensive, rigorous, lengthy, and uncertain than the 510(k) premarket notification process and de novo classification process and requires proof of the safety and effectiveness of the device to the FDA’s satisfaction. As part of the PMA review, the FDA will typically inspect the manufacturer’s facilities for compliance with Quality System Regulation (“QSR”) requirements, which impose elaborate testing, control, documentation and other quality assurance procedures. The FDA has a user fee goal to review a PMA in 180 calendar review days if the submission does not require advisory committee input, or 320 review days if the submission does require advisory committee input. During the process, the FDA may issue a major deficiency letter, which stops the review clock. The applicant has up to 180 days to respond. Therefore, the total review time could be up to 360 days, if the submission does not require advisory committee input, or 500 days if the submission does require advisory committee input, and in practice may be longer. The COVID-19 pandemic has significantly increased the FDA’s workload because of the need to review emergency use authorization requests for IVDs and other regulated products, which has delayed review timelines for some non-COVID-19 products.
If the FDA’s evaluation of the PMA application is favorable, the FDA will issue a PMA for the approved indications, which can be more limited than those originally sought by the manufacturer. The PMA can include post-approval conditions that the FDA believes necessary to ensure the safety and effectiveness of the device including, among other things, restrictions on labeling, promotion, sale and distribution or a requirement for postmarket surveillance or completion of postmarket studies. Failure to comply with the conditions of approval can result in material adverse enforcement action, including the loss or withdrawal of the approval and/or placement of restrictions on the sale of the device until the conditions are satisfied.
Even after approval of a PMA, a new PMA or PMA supplement may be required in the event of a modification to the device, its labeling or its manufacturing process. Supplements to a PMA may require the submission of the same type of information required for an original PMA, except that the supplement is generally limited to that information needed to support the proposed change from the product covered by the original PMA.
Clinical Trials
Generally, at least one clinical trial is required to support a PMA application. Clinical studies also may be required for de novo classification or a 510(k) premarket notification. Clinical trials may also be conducted or continued to satisfy post-approval requirements for devices with PMAs. For significant risk investigational device studies, the FDA regulations require that human clinical investigations conducted in the U.S. be subject to an approved investigational device exemption (“IDE”). An IDE application is considered approved 30 days after it has been received by the FDA, unless the FDA otherwise informs the sponsor prior to that time that the IDE is approved, approved with conditions, or disapproved. A nonsignificant risk investigational device study does not require FDA approval of an IDE. Some types of device studies, including many IVD studies, are exempt from IDE requirements altogether.
Clinical trials must be conducted in accordance with good clinical practice (“GCP”) requirements contained in federal regulations and in international guidelines. Clinical trials, for both significant and nonsignificant risk devices, as well as exempt studies, must be approved by an institutional review board (an “IRB”), an appropriately constituted group that has been formally designated to review and monitor biomedical research involving human subjects and which has the authority to approve, require modifications in, or disapprove research to protect the rights, safety, and welfare of the human research subject.
The FDA may order the temporary, or permanent, discontinuation of a clinical trial at any time, or impose other sanctions, if it believes that the clinical trial either is not being conducted in accordance with the FDA requirements or presents an unacceptable risk to the clinical trial patients. An IRB may also require the clinical trial it has approved to be halted, either temporarily or permanently, for failure to comply with the IRB’s requirements, or may impose other conditions or sanctions.
Although the QSR does not fully apply to investigational devices, the requirement for controls on design and development does apply. The sponsor also must manufacture the investigational device in conformity with the quality controls described in the IDE application and any conditions of IDE approval that the FDA may impose with respect to manufacturing.
Postmarket Requirements
After a device is placed on the market, numerous general regulatory controls apply. These include: the QSR, labeling regulations, medical device reporting regulations (which require that manufacturers report to the FDA if their device may have caused or contributed to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if it were to recur), and reports of corrections and removals regulations (which require manufacturers to report recalls or removals and field corrections to the FDA if initiated to reduce a risk to health posed by the device or to remedy a violation of the FDCA). Failure to properly identify reportable events or to file timely reports, as well as failure to address each of the observations to the FDA’s satisfaction, can subject a manufacturer to warning letters, recalls, or other sanctions and penalties.
Advertising, marketing and promotional activities for devices are also subject to FDA oversight and must comply with the statutory standards of the FDCA, and the FDA’s implementing regulations.
Manufacturers of medical devices are permitted to promote products solely for the uses and indications set forth in the approved or cleared product labeling. A number of enforcement actions have been taken against manufacturers that promote products for “off-label” uses (i.e., uses that are not described in the approved or cleared labeling).
Violations of the FDCA relating to inappropriate promotion of medical devices may also lead to investigations alleging violations of federal and state healthcare fraud and abuse and other laws, as well as state consumer protection laws.
| 74 |
For a PMA or Class II 510(k) or de novo device, the FDA also may require postmarketing testing, surveillance, or other measures to monitor the effects of an approved or cleared product. The FDA may place conditions on a PMA-approved device that could restrict the distribution or use of the product. In addition, quality control, manufacture, packaging, and labeling procedures must continue to conform to the QSR after approval and clearance, and manufacturers are subject to periodic inspections by the FDA. Accordingly, manufacturers must continue to expend time, money, and effort in the areas of production and quality control to maintain compliance with the QSR and other applicable regulatory requirements. The FDA may withdraw product approvals or recommend or require product recalls if a company fails to comply with regulatory requirements.
Therapeutic Products
FDA Approval Process
In the United States, therapeutic products are subject to extensive regulation by the FDA. The FDCA and other federal and state statutes and regulations, govern, among other things, the research, development, testing, manufacture, storage, recordkeeping, approval, labeling, promotion and marketing, distribution, post-approval monitoring and reporting, sampling, and import and export of pharmaceutical products. Failure to comply with applicable U.S. requirements may subject a company to a variety of administrative or judicial sanctions, such as clinical hold, FDA refusal to approve pending new drug applications (“NDAs”), warning or untitled letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties, and criminal prosecution.
Development for a new therapeutic product in the United States typically involves preclinical laboratory and animal tests, the submission to the FDA of an investigational new drug application (an “IND”), which must become effective before clinical testing may commence, and adequate and well-controlled clinical trials to establish the safety and effectiveness of the drug for each indication for which FDA approval is sought. Satisfaction of FDA premarket approval requirements typically takes many years and the actual time required may vary substantially based upon the type, complexity, and novelty of the product or disease.
Preclinical tests include laboratory evaluation of product chemistry, formulation, and toxicity, as well as animal trials to assess the characteristics and potential safety and efficacy of the product. The conduct of the preclinical tests must comply with federal regulations and requirements, including Good Laboratory Practices. The results of preclinical testing are submitted to the FDA as part of an IND along with other information, including information about product chemistry, manufacturing and controls, a general investigational plan, and a proposed clinical trial protocol. Long-term preclinical tests, such as tests of reproductive toxicity and carcinogenicity in animals, may continue after the IND is submitted. A 30-day waiting period after the submission of each IND is required prior to the commencement of clinical testing in humans. If the FDA has neither commented on nor questioned the IND within this 30-day period, the clinical trial proposed in the IND may begin. If the IND is placed on clinical hold, the sponsor must resolve any issues to the satisfaction of the FDA before the clinical hold is lifted and the clinical trial may proceed.
Clinical trials involve the administration of the investigational drug to healthy volunteers or patients under the supervision of a qualified investigator. Clinical trials must be conducted: (i) in compliance with federal regulations; (ii) in compliance with GCP requirements; and (iii) under protocols detailing the objectives of the trial, the parameters to be used in monitoring safety, and the effectiveness criteria to be evaluated. Each protocol involving testing on U.S. patients and subsequent protocol amendments must be submitted to the FDA as part of the IND.
The FDA may order the temporary or permanent discontinuation of a clinical trial at any time, or impose other sanctions, if it believes that the clinical trial either is not being conducted in accordance with FDA regulations or presents an unacceptable risk to the clinical trial patients. Imposition of a clinical hold may be full or partial. The study protocol and informed consent information for patients in clinical trials must also be submitted to an IRB for approval. The IRB will also monitor the clinical trial until completed. An IRB may also require the clinical trial at the site to be halted, either temporarily or permanently, for failure to comply with the IRB’s requirements, or may impose other conditions. Additionally, some clinical trials are overseen by an independent group of qualified experts organized by the clinical trial sponsor, known as a data safety monitoring board or committee. This group provides authorization for whether a trial may move forward at designated checkpoints based on access to certain data from the trial.
Clinical trials to support NDAs for marketing authorization are typically conducted in three sequential phases, which may overlap or be combined. In Phase 1, the initial introduction of the drug into patients, the product is tested to assess safety, dosage tolerance, metabolism, pharmacokinetics, pharmacological actions, side effects associated with drug exposure, and to obtain early evidence of a treatment effect if possible. Phase 2 usually involves trials in a limited patient population to determine the effectiveness of the drug for a particular indication, determine optimal dose and regimen, and to identify common adverse effects and safety risks. If a compound demonstrates evidence of effectiveness and an acceptable safety profile in Phase 2 evaluations, Phase 3 trials are undertaken to obtain additional information about clinical effects and confirm efficacy and safety in a larger number of patients, typically at geographically dispersed clinical trial sites, to permit the FDA to evaluate the overall benefit-risk relationship of the drug and to provide adequate information for the labeling of the product. In most cases, the FDA requires two adequate and well-controlled Phase 3 clinical trials to demonstrate the safety and efficacy of the drug. In rare instances, a single Phase 3 trial may be sufficient when either (1) the trial is a large, multicenter trial demonstrating internal consistency and a statistically very persuasive finding of a clinically meaningful effect on mortality, irreversible morbidity or prevention of a disease with a potentially serious outcome and confirmation of the result in a second trial would be practically or ethically impossible or (2) the single trial is supported by other confirmatory evidence. Approval on the basis of a single trial may be subject to a requirement for additional post-approval studies.
These phases may overlap or be combined. For example, a Phase 1/2 clinical trial may contain both a dose escalation stage and a dose-expansion stage, the latter of which may confirm tolerability at the recommended dose for expansion in future clinical trials (as in traditional Phase 1 clinical trials) and provide insight into the anti-tumor effects of the investigational therapy in selected subpopulation(s). Typically, during the development of oncology therapies, all subjects enrolled in Phase 1 clinical trials are disease-affected patients and, as a result, considerably more information on clinical activity may be collected during such trials than during Phase 1 clinical trials for non-oncology therapies.
In addition, the manufacturer of an investigational drug in a Phase 2 or Phase 3 clinical trial for a serious or life-threatening disease is required to make available, such as by posting on its website, its policy on evaluating and responding to requests for expanded access to such investigational drug.
While the IND is active, progress reports summarizing the results of the clinical trials and nonclinical studies performed since the last progress report, among other information, must be submitted at least annually to the FDA, and written IND safety reports must be submitted to the FDA and investigators for serious and unexpected suspected adverse events, findings from other studies suggesting a significant risk to humans exposed to the same or similar drugs, findings from animal or in vitro testing suggesting a significant risk to humans, and any clinically important increased incidence of a serious suspected adverse reaction compared to that listed in the protocol or investigator brochure.
| 75 |
After completion of the required clinical testing, an NDA is prepared and submitted to the FDA. FDA approval of the NDA is required before marketing and distribution of the product may begin in the United States. The NDA must include the results of all preclinical, clinical, and other testing and a compilation of data relating to the product’s pharmacology, chemistry, manufacture, and controls. The cost of preparing and submitting an NDA is substantial. The submission of most NDAs is additionally subject to a substantial application user fee. Under an approved NDA, the applicant is also subject to an annual program fee. These fees typically increase annually. The FDA has 60 days from its receipt of an NDA to determine whether the application will be filed based on the FDA’s determination that it is adequately organized and sufficiently complete to permit substantive review. Once the submission is filed, the FDA begins an in-depth review. The FDA has agreed to certain performance goals to complete the review of NDAs. Most applications are classified as Standard Review products that are reviewed within ten months of the date the FDA files the NDA; applications classified as Priority Review are reviewed within six months of the date the FDA files the NDA. An NDA can be classified for Priority Review when the FDA determines the drug has the potential to treat a serious or life-threatening condition and, if approved, would be a significant improvement in safety or effectiveness compared to available therapies. The review process for both standard and priority reviews may be extended by the FDA for three or more additional months to consider certain late-submitted information, or information intended to clarify information already provided in the NDA submission.
The FDA may also refer applications for novel products, as well as products that present difficult questions of safety or efficacy, to be reviewed by an advisory committee—typically a panel that includes clinicians, statisticians and other experts—for review, evaluation, and a recommendation as to whether the NDA should be approved. The FDA is not bound by the recommendation of an advisory committee, but generally follows such recommendations. Before approving an NDA, the FDA will typically inspect one or more clinical sites to assure compliance with GCP. Additionally, the FDA will inspect the facility or the facilities at which the drug product is manufactured. The FDA will not approve the product unless compliance with current good manufacturing practices (“cGMP”) is satisfactory. After the FDA evaluates the NDA and completes any clinical and manufacturing site inspections, it issues either an approval letter or a complete response letter. A complete response letter generally outlines the deficiencies in the NDA submission and may require substantial additional testing, or information, in order for the FDA to reconsider the application for approval. If, or when, those deficiencies have been addressed to the FDA’s satisfaction in a resubmission of the NDA, the FDA will issue an approval letter. The FDA has committed to reviewing such resubmissions in two or six months depending on the type of information included. An approval letter authorizes commercial marketing and distribution of the drug with specific prescribing information for specific indications. As a condition of NDA approval, the FDA may require a risk evaluation and mitigation strategy (a “REMS”) to help ensure that the benefits of the drug outweigh the potential risks to patients. A REMS can include medication guides, communication plans for healthcare professionals, and elements to assure a product’s safe use (“ETASU”). ETASU can include, but are not limited to, special training or certification for prescribing or dispensing the product, dispensing the product only under certain circumstances, special monitoring, and the use of patient-specific registries. The requirement for a REMS can materially affect the potential market and profitability of the product. Moreover, the FDA may require substantial post-approval testing and surveillance to monitor the product’s safety or efficacy.
Once granted, product approvals may be withdrawn if compliance with regulatory standards is not maintained, or problems are identified following initial marketing. Changes to some of the conditions established in an approved NDA, including changes in indications, product labeling, manufacturing processes or facilities, require submission and FDA approval of a new NDA, or supplement to an approved NDA, before the change can be implemented. An NDA supplement for a new indication typically requires clinical data similar to that in the original application, and the FDA uses the same procedures and actions in reviewing NDA supplements as it does in reviewing original NDAs.
Disclosure of Clinical Trial Information
Sponsors of clinical trials of FDA-regulated products, including drugs products, are required to register and disclose certain clinical trial information on the website www.clinicaltrials.gov. Information related to the product, patient population, phase of investigation, trial sites and investigators, and other aspects of a clinical trial are then made public as part of the registration. Sponsors are also obligated to disclose the results of their clinical trials after completion. Disclosure of the results of clinical trials can be delayed in certain circumstances for up to two years after the date of completion of the trial. Competitors may use this publicly available information to gain knowledge regarding the progress of clinical development programs as well as clinical trial design.
Pediatric Information
Under the Pediatric Research Equity Act (the “PREA”), NDAs or supplements to NDAs must contain data to assess the safety and effectiveness of the drug product for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the drug product is safe and effective. The FDA may grant full or partial waivers, or deferrals, for submission of data. Unless otherwise required by regulation, PREA does not apply to any drug product with orphan product designation except a product with a new active ingredient that is a molecularly targeted cancer product intended for the treatment of an adult cancer and directed at a molecular target determined by the FDA to be substantially relevant to the growth or progression of a pediatric cancer.
The Best Pharmaceuticals for Children Act (the “BPCA”) provides a six-month extension of any patent or non-patent exclusivity for a drug if certain conditions are met. Conditions for exclusivity include the FDA’s determination that information relating to the use of a new drug in the pediatric population may produce health benefits in that population, the FDA making a written request for pediatric studies, and the applicant agreeing to perform, and reporting on, the requested studies within the statutory timeframe. Applications under the BPCA are treated as priority applications, with all of the benefits that designation confers.
Post-Approval Requirements
Once an NDA is approved, a product will be subject to certain post-approval requirements. For instance, the FDA closely regulates the post-approval marketing and promotion of drugs, including standards and regulations for direct-to-consumer advertising, off-label promotion, industry-sponsored scientific and educational activities, and promotional activities involving the Internet. A drug may be marketed only for the approved indications and in accordance with the provisions of the approved labeling.
Adverse event reporting and submission of periodic safety summary reports is required following FDA approval of an NDA. The FDA also may require postmarket testing, known as Phase 4 testing, REMS, and surveillance to monitor the effects of an approved product, or the FDA may place conditions on an approval that could restrict the distribution or use of the product. In addition, quality control, product manufacture, packaging, and labeling procedures must continue to conform to cGMP after approval. Drug manufacturers and certain of their subcontractors are required to register their establishments with the FDA and certain state agencies.
| 76 |
Registration with the FDA subjects entities to periodic unannounced inspections by the FDA, during which the agency inspects a drug product’s manufacturing facilities to assess compliance with cGMP. Accordingly, manufacturers must continue to expend time, money, and effort in the areas of production and quality-control to maintain compliance with cGMP. Regulatory authorities may withdraw product approvals or request product recalls if a company fails to comply with required regulatory standards, if it encounters problems following initial marketing, or if previously unrecognized problems are subsequently discovered.
European Union
A medical device or diagnostic test must be CE marked to be sold in the EU. The IVDR defines the necessary pre-conditions that must be fulfilled to CE mark an IVD test or in vitro medical device in the EU. The manufacture of the test and/or device must fulfill all applicable regulatory requirements in the IVDR. Objective evidence of fulfilment of these requirements must be provided by the manufacturer prior to placing a test on the EU market. The manufacturer is required to establish a Quality Management System (“QMS”) as well as processes for manufacturing, importing, distribution, post-market surveillance, and vigilance. Regulations also require that the product is fully documented. In addition, it is likely that our test is classified in a risk class that requires a review by an external party, a Notified Body, prior to placing the test on the EU market. This process is expected to require an additional 6-12 months after required documents and systems are in place. There currently is a general shortage in the EU of available Notified Bodies designated for IVDR devices. Further, we will need to contract a European Authorized Representative (“EAR”) that acts as the Company’s legal representative in the EU. Medical devices also must be registered with the competent authority in the country in which it is based. In addition to the CE mark and the registration done by the EAR, there is a need for an administrative national notification with certain member states of the EU.
European Data Collection
The collection and use of personal data (including health data) in the European Economic Area (the “EEA”) are governed by the EU General Data Protection Regulations (the “EU GDPR”) and national implementing legislation in EEA Member States. The EU GDPR applies to any company established in the EEA and to companies established outside the EEA that process personal data in connection with the offering of goods or services to data subjects in the EEA or the monitoring of the behavior of data subjects in the EEA. The EU GDPR establishes stringent requirements applicable to the processing of personal data, including strict requirements relating to the validity of consent of data subjects, expanded disclosures about how personal data is used, requirements to conduct data protection impact assessments for “high risk” processing, limitations on retention of personal data, special provisions for “special categories of personal data” including health and genetic information of data subjects, mandatory data breach notification (in certain circumstances), “privacy by design” requirements, and direct obligations on service providers acting as processors. The EU GDPR also prohibits the international transfer of personal data from the EEA to countries outside of the EEA unless made to a country deemed to have adequate data privacy laws by the European Commission or a data transfer mechanism has been put in place. Failure to comply with the requirements of the EU GDPR and the related national data protection laws of the EEA States may result in fines up to 20 million euros or 4% of a company’s global annual revenues for the preceding financial year, whichever is higher. Moreover, the EU GDPR affords various data protection rights to individuals (i.e., the right to erasure of personal data) in certain circumstances, and the ability for data subjects to claim material and non-material damages resulting from infringements of the EU GDPR. Given the breadth and depth of changes in data protection obligations, maintaining compliance with the EU GDPR will require significant time, resources, and expense, and we may be required to put in place additional mechanisms ensuring compliance with the evolving data protection rules. This may be onerous and adversely affect our business, financial condition, results of operations and prospects.
Rest of the World Regulation
For other countries outside of the EU (or in some cases, EEA) and the United States, such as China, Southeast Asia, and Australia, the requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country. Additionally, the clinical trials must be conducted in accordance with GCP requirements and the applicable regulatory requirements, and the ethical principles that have their origin in the Declaration of Helsinki.
If we fail to comply with applicable foreign regulatory requirements, we may be subject to, among other things, fines, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions, and criminal prosecution.
Human Capital
We place significant emphasis on the recruitment, development, and retention of our employees who include award-winning scientists dedicated to advancing scientific discovery from bench to bedside. Of our 17 employees, all of whom are employed full-time, one holds an M.D. and six hold Ph.Ds in biology or medicinal chemistry. Approximately nine employees are engaged in research and development and approximately five in sales or general administration.
Our Executive Vice President and Chief Medical and Science Officer, Vivienne Rebel, holds an M.D. and Ph.D. Business development is led by our Chief Operating Officer, Xavier Reveles, who has 25 years of experience as a clinical geneticist skilled in the creation and management of CLIA clinical laboratories, coding, and CPT reimbursement valuations. Mr. Reveles is board certified by the American Society of Clinical Pathology as a clinical specialist in cytogenetics who has successfully launched multiple diagnostics and commercial laboratories. Our innovative and collaborative culture is in part responsible for the high degree of retention and professional advancement. Outside partnerships and collaborations that advance business and scientific research are encouraged, allowing us to multiply workforce efforts without expending significant capital.
| 77 |
BIOAFFINITY TECHNOLOGIES, INC.
UNAUDITED PRO FORMA COMBINED FINANCIAL STATEMENTS
On September 18, 2023, Precision Pathology Laboratory Services, LLC (“PPLS”), a Texas limited liability company and wholly owned subsidiary of bioAffinity Technologies, Inc. (“bioAffinity”), entered into an Asset Purchase Agreement (the “Asset Purchase Agreement”) with Dr. Roby P. Joyce, M.D. (“Owner”) and Village Oaks Pathology Services, P.A. (the “Seller”) pursuant to which PPLS purchased the non-medical assets of the Seller (the “Acquisition”). In addition, PPLS will provide certain management services to the Seller in all clinical pathology laboratory services, administrative, and non-medical services for pathologists to support community-based pathology medical groups. Pursuant to the Asset Purchase Agreement, PPLS paid at the Closing a cash payment of $2,500,000 to Seller ($1,822,630) and debt balances owed ($370,370) at the time of the Acquisition, and paid into an escrow account $350,000 to satisfy contingent and non-contingent post-closing obligations and issued 564,972 shares of bioAffinity’s common stock to Seller having a value of $1,000,000.
The following unaudited pro forma condensed combined financial statements have been prepared to give effect to the Acquisition. These unaudited pro forma condensed combined financial statements are derived from the historical consolidated financial statements of the bioAffinity and PPLS. These financial statements have been adjusted as described in the notes to the unaudited pro forma condensed combined financial statements.
The unaudited pro forma condensed combined balance sheet combines the historical consolidated balance sheets of the bioAffinity and PPLS, has been prepared assuming the Acquisition closed on December 31, 2022, and includes preliminary adjustments to reflect the events that are directly attributable to the Acquisition and factually supportable. In addition, the unaudited pro forma condensed combined statement of operations for the year ended December 31, 2022 and the six months ended June 20, 2023, combines the historical consolidated statements of operations of the bioAffinity and PPLS as if the Acquisition has occurred on January 1, 2022 and has also been adjusted to give effect to pro forma events that are directly attributable to the Acquisition, factually supportable and expected to have a continuing impact on the combined results.
bioAffinity has prepared the unaudited pro forma combined condensed financial statements based on available information using assumptions that it believes are reasonable. These pro forma financial statements are being provided for informational purposes only and do not claim to represent bioAffinity’s actual financial position or results of operations had the Acquisition occurred on that date specified nor do they project bioAffinity’s results of operations or financial position for any future period or date. The actual results reported by the combined company in periods following the Acquisition may differ significantly from these unaudited pro forma combined condensed financial statements for a number of reasons. The pro forma financial statements do not account for the cost of any restructuring activities or synergies resulting from the Acquisition or other costs relating to the integration of the two companies, or other historical acquisitions that were undertaken by bioAffinity.
The unaudited pro forma combined condensed financial statements were prepared using the acquisition method of accounting as outlined in Financial Accounting Standards Board Accounting Standards Codification (“ASC”) 805, Business Combinations, with bioAffinity considered the acquiring company. Based on the acquisition method of accounting, the consideration paid for the non-medical assets of the Seller is allocated to its assets and liabilities based on their fair value as of the date of the completion of the Acquisition. The purchase price allocation and valuation is based on preliminary estimates, subject to final adjustments and provided for informational purposes only.
These unaudited pro forma combined condensed financial statements should be read in conjunction with the bioAffinity’s historical consolidated financial statements and accompanying notes included in bioAffinity’s Annual Report on Form 10-K for the year ended December 31, 2022.
| 78 |
BIOAFFINITY TECHNOLOGIES, INC.
UNAUDITED PRO FORMA CONDENSED COMBINED BALANCE SHEET
AS OF JUNE 30, 2023
| Historical | Pro forma | Pro forma | ||||||||||||||
| bioAffinity | PPLS1 | Adjustments | Combined | |||||||||||||
| ASSETS | ||||||||||||||||
| Current assets: | ||||||||||||||||
| Cash and cash investments | $ | 8,279,182 | $382,648 | (1) | $ | (2,500,000 | ) | $ | 6,161,830 | |||||||
| Accounts and other receivables, net | 90,232 | 1,330,792 | 1,421,024 | |||||||||||||
| Inventory | 10,101 | - | 10,101 | |||||||||||||
| Prepaid assets | 279,687 | 9,316 | 289,003 | |||||||||||||
| Total current assets | 8,659,203 | 1,722,756 | (2,500,000 | ) | 7,881,958 | |||||||||||
| Property and Equipment, net of accumulated depreciation | 207,377 | 339,978 | 547,355 | |||||||||||||
| Operating lease right-of-use asset, net | - | 445,599 | 445,599 | |||||||||||||
| Finance lease right-of-use asset, net | - | 1,183,652 | 1,183,652 | |||||||||||||
| Goodwill | - | - | (2) | 1,990,520 | 1,990,520 | |||||||||||
| Other assets | 6,920 | 8,000 | 14,920 | |||||||||||||
| Total non-current assets | 214,297 | 1,977,229 | 1,990,520 | 4,182,046 | ||||||||||||
| Total assets | $ | 8,873,499 | $ | 3,699,985 | $ | (509,480 | ) | $ | 12,064,004 | |||||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY | ||||||||||||||||
| Current liabilities: | ||||||||||||||||
| Accounts payable | $ | 174,404 | $ | 65,644 | $ | 240,048 | ||||||||||
| Accrued expenses | 515,663 | 247,129 | 762,792 | |||||||||||||
| Unearned Revenue | 42,750 | - | 42,750 | |||||||||||||
| Notes payable, current portion | - | 19,506 | (3) | (19,506 | ) | - | ||||||||||
| Operating lease liability, current portion | - | 101,570 | 101,570 | |||||||||||||
| Finance lease liability, current portion | - | 393,626 | 393,626 | |||||||||||||
| Short-term loan | 42,334 | 198,000 | (3) | (198,000 | ) | 42,334 | ||||||||||
| Total current liabilities | 775,152 | 1,025,475 | (217,506 | ) | 1,583,120 | |||||||||||
| Operating lease liability, net of current portion | - | 350,619 | 350,619 | |||||||||||||
| Finance lease liability, net of current portion | - | 1,031,917 | 1,031,917 | |||||||||||||
| Notes payable, net of long-term portion | - | 112,424 | (3) | (112,424 | ) | - | ||||||||||
| Total non-current liabilities | - | 1,494,960 | - | 1,382,536 | ||||||||||||
| Total liabilities | 775,152 | 2,520,435 | (329,930 | ) | 2,965,656 | |||||||||||
| Stockholders (deficit) equity: | ||||||||||||||||
| Common stock | 59,887 | 5 | (4) | 3,955 | 64,518 | |||||||||||
| (5) | (5 | ) | ||||||||||||||
| (6) | 676 | |||||||||||||||
| Additional paid-in capital | 47,978,892 | - | (4) | 996,045 | 49,145,303 | |||||||||||
| (6) | 170,366 | |||||||||||||||
| Accumulated (deficit) / earnings | (39,940,431 | ) | 1,179,545 | (5) | (1,179,545 | ) | (40,111,473 | ) | ||||||||
| (6) | (171,042 | ) | ||||||||||||||
| Total stockholders’ (deficit) equity | 8,098,348 | 1,179,550 | (179,550 | ) | 9,098,348 | |||||||||||
| Total liabilities and stockholders’ equity | $ | 8,873,499 | $ | 3,699,985 | $ | (509,480 | ) | $ | 12,064,004 | |||||||
| 79 |
BIOAFFINITY TECHNOLOGIES, INC.
UNAUDITED PRO FORMA COMBINED STATEMENT OF OPERATIONS
For Full Year Ended December 31, 2022
| Historical | Pro forma | Pro forma | ||||||||||||||
| bioAffinity | PPLS | Adjustments | Combined | |||||||||||||
| Net revenues | $ | 4,803 | $ | 6,858,212 | $ | 6,863,015 | ||||||||||
| Less: cost of sales | (467 | ) | - | (467 | ) | |||||||||||
| Gross Profit | 4,336 | $ | 6,858,212 | 6,862,548 | ||||||||||||
| Operating expenses: | ||||||||||||||||
| Research and development | 1,142,777 | - | 1,142,777 | |||||||||||||
| Clinical development | 145,546 | - | 145,546 | |||||||||||||
| Selling, General and administrative | 2,716,889 | 7,184,802 | 9,901,691 | |||||||||||||
| Depreciation and Amortization | 10,182 | 544,217 | 554,399 | |||||||||||||
| Total operating expenses | 4,015,394 | 7,729,019 | 11,744,413 | |||||||||||||
| Loss from operations | (4,011,058 | ) | (870,807 | ) | (4,881,865 | ) | ||||||||||
| Other income (expense): | ||||||||||||||||
| Interest income | 46,708 | 9,192 | 55,900 | |||||||||||||
| Interest expense | (2,532,640 | ) | (63,308 | ) | (2,595,948 | ) | ||||||||||
| Other Income | - | 8,775 | 8,775 | |||||||||||||
| Gain on extinguishment of debt | 212,258 | 503,950 | 716,208 | |||||||||||||
| Unrealized gain (loss) on investments | (1,866,922 | ) | (49,434 | ) | (1,916,356 | ) | ||||||||||
| Net loss before income taxes | (8,151,654 | ) | (461,632 | ) | (8,613,286 | ) | ||||||||||
| Income tax expense | (2,459 | ) | - | (2,459 | ) | |||||||||||
| Net income (loss) | $ | (8,154,113 | ) | $ | (461,632 | ) | $ | (8,615,745 | ) | |||||||
| Net loss per common share, basic and diluted | $ | (1.81 | ) | $ | (1.70 | ) | ||||||||||
| Weighted average common shares outstanding, basic, and diluted | 4,498,964 | (4) | 564,972 | 5,063,936 | ||||||||||||
| 80 |
BIOAFFINITY TECHNOLOGIES, INC.
UNAUDITED PRO FORMA COMBINED STATEMENT OF OPERATIONS
For Six Months Ended June 30, 2023
| Historical | Pro forma | Pro forma | ||||||||||||||
| bioAffinity | PPLS | Adjustments | Combined | |||||||||||||
| Net revenues | $ | 20,659 | $ | 3,610,549 | $ | 3,631,208 | ||||||||||
| Less: cost of sales | (1,322 | ) | - | (1,322 | ) | |||||||||||
| Gross Profit | 19,337 | 3,610,549 | 3,629,886 | |||||||||||||
| Operating expenses: | ||||||||||||||||
| Research and development | 704,741 | - | 704,741 | |||||||||||||
| Clinical development | 54,888 | - | 54,888 | |||||||||||||
| Selling, General and administrative | 2,552,792 | 3,633,108 | 6,185,900 | |||||||||||||
| Depreciation and Amortization | 43,236 | 430,844 | 474,080 | |||||||||||||
| Total operating expenses | 3,355,657 | 4,063,952 | 7,419,609 | |||||||||||||
| Loss from operations | (3,336,320 | ) | (453,403 | ) | (3,789,723 | ) | ||||||||||
| Other income (expense): | ||||||||||||||||
| Interest income | 82,778 | 4,880 | 87,658 | |||||||||||||
| Interest expense | (3,015 | ) | (57,777 | ) | (60,792 | ) | ||||||||||
| Other Income | - | 5,149 | 5,149 | |||||||||||||
| Unrealized gain (loss) on investments | - | 8,131 | 8,131 | |||||||||||||
| Net loss before income taxes | (3,256,557 | ) | (493,020 | ) | (3,749,577 | ) | ||||||||||
| Income tax expense | (16,406 | ) | - | (16,406 | ) | |||||||||||
| Net income (loss) | $ | (3,272,963 | ) | $ | (493,020 | ) | $ | (3,765,983 | ) | |||||||
| Net loss per common share, basic and diluted | $ | (0.38 | ) | $ | (0.42 | ) | ||||||||||
| Weighted average common shares outstanding, basic, and diluted | 8,477,656 | (4) | 564,972 | 9,042,628 | ||||||||||||
| 81 |
NOTES TO UNAUDITED PRO FORMA CONDENSED COMBINED FINANCIAL STATEMENTS
Basis of Presentation
The unaudited pro forma condensed combined balance sheet as of December 31, 2022 combines the historical consolidated balance sheets of bioAffinity and PPLS and has been prepared as if the Acquisition had occurred on December 31, 2022. The unaudited pro forma combined statement of operations for the year ended December 31, 2022 and the six months ended June 30, 2023 combines the historical consolidated statement of operations of bioAffinity and PPLS and has been prepared has been prepared as if the Acquisition closed on January 1, 2022. The unaudited pro forma condensed combined financial statements have also been adjusted to give effect to pro forma events that are directly attributable to the Acquisition, factually supportable and expected to have a continuing impact on the combined results.
The Acquisition was accounted for under the acquisition method of accounting in accordance with ASC 805, Business Combinations. Under the acquisition method, the total estimated purchase price, or consideration transferred, is measured at the Acquisition closing date. The assets of the Seller have been measured based on various preliminary estimates using assumptions that bioAffinity’s management believes are reasonable utilizing information currently available. As such, the net book value of the assets (equipment, property and leases) at Acquisition are estimated to be materiality in line with estimated fair value. Therefore, no pro forma adjustments have been made to the acquired assets.
These pro forma financial statements are being provided for informational purposes only and do not claim to represent bioAffinity’s actual financial position or results of operations had the Acquisition occurred on that date specified nor do they project bioAffinity’s results of operations or financial position for any future period or date. The actual results reported by the combined company in periods following the Acquisition may differ significantly from these unaudited pro forma combined condensed financial statements for a number of reasons. The pro forma financial statements do not account for the cost of any restructuring activities or synergies resulting from the Acquisition or other costs relating to the integration of the two companies, or other historical acquisitions that were undertaken by bioAffinity.
Purchase Price
The unaudited pro forma condensed combined financial information reflects the purchase price as follows:
| Cash | $ | 2,500,000 | ||
| bioAffinity common stock issued (564,972 @ $1.77) | 1,000,000 | |||
| Purchase Price | $ | 3,500,000 |
Purchase Price Allocation
| Cash | $ | 382,648 | ||
| Receivables | 1,330,792 | |||
| Prepaids | 9,316 | |||
| PP&E | 339,978 | |||
| Right to use lease asset | 1,629,251 | |||
| Deposits | 8,000 | |||
| Payables | (65,644 | ) | ||
| Accrued Expenses | (247,129 | ) | ||
| Right to use lease liability | (1,877,732 | ) | ||
| Goodwill | 1,990,520 | |||
| $ | 3,500,000 |
Pro forma adjustments
The pro forma adjustments included in the unaudited pro forma condensed combined financial statements are as follows:
| (1) | Cash: Reflects $2.5 million in cash consideration paid to the Seller. | |
| (2) | Goodwill: Adjustments to record goodwill resulting from the Acquisition. Goodwill is not amortized but rather is assessed for impairment at least annually or more frequently whenever events or circumstances indicate that goodwill might be impaired. | |
| (3) | Debt: Adjustments to reflect the line of credit, notes payable, current portion, and notes payable, net of current portion of $198 thousand, $20 thousand, and $112 thousand, respectively, paid off by the Seller at closing. | |
| (4) | Stock Issuance: Reflects the effect of 564,972 in common stock shares issuance to the Seller at a stock price of $1.77 at par value of $0.007. | |
| (5) | Stockholders’ Equity: Adjustment to eliminate the Seller’s historical stockholders’ equity. | |
| (6) | bioAffinity employee and board stock issuance: Reflects stock issuance of 71,715 restricted shares to board of directs on 7/1/2022, 16,605 restricted stock issued before 6/30/2023 that has since vested, and 8,226 restricted shares to Peter Connor on 7/1/2022 (2,732), 8/1/2022 (2,717), and 9/1/2022 (2,777) at a par value of $0.007. |
| 82 |
MANAGEMENT
The following is a list of our directors and executive officers as of September 20, 2023.
| Name | Age | Position(s) | ||
| Maria Zannes, JD | 67 | President, CEO, and Director | ||
| Vivienne Rebel, M.D., Ph.D | 58 | Executive Vice President and Chief Science and Medical Officer | ||
| Michael Dougherty | 45 | Chief Financial Officer | ||
| Xavier Reveles | 54 | Chief Operating Officer | ||
| Timothy P. Zannes, JD | 71 | Executive Vice President, Secretary, and General Counsel | ||
| Steven Girgenti | 77 | Executive Chairman and Director | ||
| Robert Anderson | 82 | Director | ||
| Stuart Diamond | 62 | Director | ||
| Peter Knight | 72 | Director | ||
| Mohsin Meghji | 58 | Director | ||
| Gary Rubin | 67 | Director | ||
| Roby P. Joyce, M.D. | 75 | Director |
Biographical Information
Maria Zannes, JD — President, Chief Executive Officer, and Director
Ms. Zannes has served as our President, Chief Executive Officer and director since 2014. She brings more than 30 years of executive-level management experience dedicated to defining and advancing company goals and overcoming obstacles impeding corporate success. Prior to her position at bioAffinity Technologies, Ms. Zannes founded The Zannes Firm focusing on strategic solutions for private industry in the medical, environmental and energy fields. In her current capacity as the Company’s CEO and President, she has built a team of award-winning scientists and executives who are advancing breakthrough oncology-focused diagnostics and therapeutics.
Ms. Zannes was President of the Energy Recovery Council, the national trade group for the $10 billion waste-to-energy industry, and General Manager of ECOS Corporation, a subsidiary of Burlington Environmental. Ms. Zannes also served as a project manager at Wheelabrator Technologies, Inc. where she led project teams that developed, negotiated, and financed the company’s renewable energy generation facilities. Ms. Zannes began her career as a journalist, working for Voice of America and the Associated Press. Before entering the business world, she served as a legislative aide specializing in energy policy and law for Congressman Charles Wilson (D-TX). She is licensed to practice law in New Mexico. She has been awarded Lifetime Achievement Awards by the American Society of Mechanical Engineers and the Earth Engineering Center Award from the WTE Council of Columbia University.
She is the co-founder of two engineering research centers at Columbia University. Ms. Zannes received her BA in Journalism from the University of New Mexico and her JD from the University of Puget Sound in Washington State. We believe Ms. Zannes should serve as a Director because of her experience as a lawyer and business woman skilled in identifying, prioritizing and managing both the risks and rewards of business opportunities and her proven record of assembling and motivating award-winning teams of professionals focused on strategic corporate growth.
Vivienne I. Rebel, M.D., Ph.D — Executive Vice President and Chief Science and Medical Officer
Dr. Rebel, a cancer (stem) cell biologist, with more than 20 years of experience in scientific research, focused on understanding the molecular events that lead to cancer development, has served as our Executive Vice President and Chief Science and Medical Officer since 2016. She received her M.D. and Ph.D from the Free University in Amsterdam, the Netherlands, and post-doctoral training at the Dana-Farber Cancer Institute, Harvard Medical School. From 2005 to 2016, she led her own research group at The University of Texas Health Science Center at San Antonio, where she first collaborated with bioAffinity. Dr. Rebel received the 2012 Cancer Therapy & Research Center Discovery of the Year Award. She is the (co)author of more than 50 publications in peer-reviewed journals.
Michael Dougherty — Chief Financial Officer
Michael Dougherty was appointed to serve as our Vice President and Chief Financial Officer (principal financial officer and principal accounting officer) effective May 1, 2023. Prior to joining the Company, Mr. Dougherty served from 2022 through April 2023 as the Chief Financial Officer of Alexa Business Domains, Amazon’s Alexa Commercial domains. From 2020 to 2022, Mr. Dougherty was Chief Financial Officer of TINT and Filestack, two software-as-a-service companies. From 2017 to 2020, Mr. Dougherty served as Chief Financial Officer for Amazon Pay, where he supported Amazon’s digital payment wallet globally. Earlier in his career Mr. Dougherty held various finance positions at Russell Investments and Medisystems Corporation. He is a certified public accountant and a Chartered Global Management Accountant by the American Institute of Certified Public Accountants.
Xavier Reveles – Chief Operating Officer
Mr. Reveles was appointed to serve as our Chief Operating Officer on September 14, 2023. He has 30 years of experience as a clinical cytogeneticist skilled in the design/concept and management of CAP CLIA clinical laboratories, coding, CPT reimbursement valuations, and the development of LDTs. Mr. Reveles is board certified by the American Society of Clinical Pathology as a clinical specialist in cytogenetics. He joined bioAffinity as Director of Operations in 2017. Prior to joining bioAffinity, Mr. Reveles created the Oncopath Laboratory – START Cancer Center (“Oncopath”) in San Antonio, Texas, and served as Laboratory Director. During his tenure at Oncopath, he commercialized eight LDTs, including bringing to market a proprietary cancer specific gene oligo array he designed for the deletions and amplifications of specific oncogenes for solid tumors. As the Director of the Cytogenetics Laboratory at UT Health San Antonio, Mr. Reveles’ research included molecular evaluation of disease progression in prostate, breast and ovarian cancer, schizophrenia, diabetes and other constitutional genetic syndromes. He was a lecturer and instructor for the UT Health Graduate, Medical, and Allied Health Schools and the director of the NCI San Antonio Cancer Institute (SACI) Genetics and Cytogenetics Core facility. After leaving academia, Mr. Reveles was a genomic specialist for CombiMatrix Diagnostics, in Irvine, California, a diagnostic biotech company where he validated pre-natal, post-natal, and cancer gene arrays for commercialization as LDTs. Mr. Reveles is (co)author of 20 publications and 6 abstracts in peer-reviewed journals and is a member of the Association for Molecular Pathology.
Timothy P. Zannes, JD — Executive Vice President, General Counsel, and Secretary
Mr. Zannes has been corporate legal counsel to both public and private biomedical firms for more than 16 years, having begun his legal career as a sole practitioner accepting criminal, business, family, and tort litigation. Prior to receiving his JD, Mr. Zannes was a court bailiff and ran his own private investigation firm after serving as an investigator for the Albuquerque City Attorney. He received his JD from the University of New Mexico School of Law and attended the New England Conservatory with studies in violin and saxophone. Mr. Zannes began his undergraduate education at The University of North Carolina where he was a student athlete on scholarship. In addition to his duties as General Counsel and Secretary, Mr. Zannes is responsible for corporate compliance and directs Human Resources. Mr. Zannes and Maria Zannes are siblings.
Steven Girgenti — Executive Chairman of the Board
Mr. Girgenti is a veteran healthcare executive with a foundation of expertise in healthcare marketing strategies, financing, and mergers and acquisitions. He has been Executive Chairman of bioAffinity Technologies, Inc. since November 2014. Mr. Girgenti was formerly CEO and co-founder of DermWorx Incorporated, a dermatology company that specialized in developing nanotechnology formulations to enhance the performance of topical drugs. He was also the founder and CEO of Healthworld Corporation, a leading global healthcare marketing services network with offices in 36 countries, until 2008. The network had more than 1,000 brand assignments from nearly 200 clients worldwide, providing strategic marketing and communications services to many of the world’s leading healthcare companies. Mr. Girgenti founded Healthworld in 1986, and under his leadership the Company made numerous acquisitions to expand and diversify the business. Healthworld went public in 1997. In 1998 and again in 1999, Business Week named Healthworld one of the “Best Small Corporations in America.” In 1999, Forbes listed Healthworld as one of the “200 Best Small Companies.” Mr. Girgenti was recognized as “Entrepreneur of the Year” by Nasdaq in 1999 and was named Med Ad News’ first “Medical Advertising Man of the Year” in 2000. In 2010, he was inducted into the Medical Advertising Hall of Fame. In addition, Mr. Girgenti is Vice Chairman of the Board of Governors for the Mt. Sinai Hospital Prostate Disease and Research Center in New York City and is on the Board of Directors for the Jack Martin Fund, a Mt. Sinai Hospital-affiliated charitable organization devoted to pediatric oncology research. He graduated from Columbia University. We believe Mr. Girgenti should serve as Executive Chairman because of his unparalleled experience in the healthcare field, particularly in marketing, and his skill in building emerging growth companies into multi-national corporations.
| 83 |
Robert Anderson — Director
Mr. Anderson has more than 50 years of broad experience in the healthcare industry in which he held executive positions at CIBA Pharmaceutical Co., Becton Dickinson and Company, Pfizer, Inc., Parke-Davis Division of Warner-Lambert Co, Schering-Plough Corp., and Centocor, Inc. Mr. Anderson was Vice President of Marketing for the Key Pharmaceuticals Division of Schering-Plough Corp. and later at Centocor, Inc. Subsequently, Mr. Anderson joined Physicians World Communications Group, the largest medical education company in the U.S. where he was Chief Operating Officer. Mr. Anderson currently is a marketing consultant to several healthcare companies. Mr. Anderson received a BA in political science from Rutgers University. We believe Mr. Anderson should serve as Director because of his experience and skill in marketing and product positioning of medical products to bioAffinity Technologies.
Stuart Diamond — Director
Mr. Diamond is the Global Chief Financial Officer for GroupM, the world’s leading media investment company responsible for over $50 billion in media investment through agencies Mindshare, MediaCom, Wavemaker, Essence and m/SIX, as well as the outcome-driven programmatic audience company, Xaxis, LLC. Before joining GroupM, Mr. Diamond was a member of the WPP plc family as the CFO for Healthworld Corporation (now called Ogilvy Health), where he took the company public and negotiated its sale to Cordiant Communications Group in 2000. He also served as CFO for National Medical Health Card Systems, Inc., a comprehensive pharmacy benefit management company. From 2008 to 2014, Mr. Diamond was the CFO for GroupM North America, where he established financial strategies and supervised all corporate accounting and financial activities for GroupM and its agencies. Earlier in his career, he held the positions of Vice President and Controller for Calvin Klein, Inc. and as Senior Vice President and CFO for Medicis Pharmaceutical Corporation. Mr. Diamond holds a BS from the State University of New York, a Master of Science, Taxation degree from Pace University, and an MBA from Fordham University. We believe Mr. Diamond should serve as a Director because of his substantial business and financial acumen to his position as Chairman of the Audit Committee and to the Board.
Peter S. Knight — Director
Mr. Knight is a Partner at Cyan Capital Partners, a fund dedicated to helping new fund managers and asset owners in the field of sustainable investing. Prior to that, he was a Founding Partner at Generation Investment Management, where he and his partners Al Gore and David Blood helped build a leading global sustainable investing firm with assets under management now exceeding $30 billion. Prior to his retirement from the firm in 2018, Mr. Knight held leadership positions within Generation IM, notably developing and overseeing the firm’s U.S. business. Prior to Generation, Mr. Knight was a Managing Director of Met West Financial, a Los Angeles-based asset management company. Mr. Knight started his career at the Antitrust Division of the U.S. Department of Justice. From 1977 to 1989, he served as the Chief of Staff to Representative and later Senator Al Gore. He served as the General Counsel of Medicis Pharmaceutical and then started his law practice where he represented the International Olympic Committee, the U.S. Olympic Committee, and numerous Fortune 500 Companies. Mr. Knight has also served in senior positions on four Presidential campaigns including serving as the Campaign Manager for President Clinton’s 1996 re-election campaign. Mr. Knight has extensive board experience in both the for-profit and nonprofit sectors. He served on a number of public company boards including Medicis Pharmaceutical, Par Pharmaceutical, EntreMed (Casi Pharmaceuticals Inc.), Healthworld Corporation, Whitman Education, Comsat, and the Schroder Mutual Fund Board complex. Mr. Knight currently serves on the boards of Generation Investment Management and Gratitude Railroad. His philanthropic efforts include serving as Chair of the Climate Museum and the board of Emergent, a nonprofit intermediary to help stop deforestation in tropical forest nations. He received a BA from Cornell University and a JD from the Georgetown Law School. We believe Mr. Knight should serve as a Director because of his considerable experience in finance and business to his position of Chairman of the Compensation Committee, as well as his expertise and skill in building new ventures into leading global firms.
Mohsin Meghji — Director
Mr. Meghji is a Managing Partner of M3 Partners L.P., a New York-based merchant banking firm. He is a nationally recognized turnaround professional with an exemplary track record of accomplishment across a wide range of industries. His 25+ year career has focused primarily on maximizing value for stakeholders. He has accomplished this through management and/or advisory roles in partnership with some of the world’s leading financial institutions, private equity, and distressed hedge fund investors. Mr. Meghji serves as Chairman of the Board of Infrastructure & Energy Alternatives, Inc., one of the country’s leading renewables-focused engineering and construction firms, which merged with a special purpose acquisition company sponsored by M3 Partners in March 2018 and is now listed on Nasdaq. Mr. Meghji’s most recent corporate role was as Executive Vice President and Head of Strategy at Springleaf Holdings, LLC as well as CEO of its captive insurance companies. Springleaf was listed on the New York Stock Exchange in late 2013. Mr. Meghji co-founded Loughlin Meghji + Company, a financial advisory firm which became one of the leading restructuring boutiques in the U.S. Earlier in his career, he spent over 12 years with Arthur Andersen & Co. in the firm’s London, Toronto, and New York offices as a Partner in the Global Corporate Finance group. He has served as a director on a number of corporate boards, including Mariner Healthcare Inc., Cascade Timberlands, LLC, Dan River, Inc., and MS Resorts. He is a director of the Equity Group International Foundation, which provides funding for underprivileged high-potential students in Kenya. Previously, he served on the boards of The Children’s Museum of Manhattan as well as HealthRight International from 2004 to 2012. In his capacity as a restructuring and financial advisory professional, Mr. Meghji has periodically served as an independent director or Chief Restructuring Officer (or in an analogous position) of companies which elected to utilize bankruptcy proceedings as a part of their financial restructuring process, and as such, he served as a director or executive officer of various companies which filed bankruptcy petitions under federal law. Mr. Meghji is a graduate of the Schulich School of Business, York University, Canada. He has previously qualified as a U.K. and Canadian Chartered Accountant as well as a U.S. Certified Turnaround Professional. We believe Mr. Meghji should serve as a Director because of his exemplary track record in maximizing stockholder value and managing companies to achieve financial and business success.
| 84 |
Roby P. Joyce, M.D. – Director
Dr. Joyce was appointed to serve on our Board of Directors on September 14, 2023. He is board-certified in anatomic and clinical pathology by the College of American Pathologists and is a Diplomat in the American Board of Pathology. He is also board-certified in neurology by the American Academy of Neurology and is a Diplomat in the American Board of Psychiatry and Neurology. Dr. Joyce founded Village Oaks in 2008. He is Medical Director and Laboratory Director of PPLS and owner of Village Oaks, the medical professional association whose pathologists provide pathology interpretation services to PPLS. In addition to his role at Village Oaks, he has served in various capacities at Northeast Methodist Hospital in San Antonio, including Chairman of the Board of Trustees and Chief of Staff of the Methodist Healthcare System. Throughout a career in pathology that spans more than 40 years, he has been a highly regarded speaker at medical and scientific conferences, has served in leadership roles on dozens of professional organizations and committees, and has served as lead or co-author of numerous scientific articles. Dr. Joyce received his medical degree from Louisiana State University, where he also received a BS in zoology. He performed his internship at Fitzsimons Army Medical Center in Denver, his residency in neurology at the Letterman Army Medical Center at the University of California Moffett Hospital in San Francisco, and his residency in pathology at Brooke Army Medical Center in San Antonio. We believe Dr. Joyce should serve as a Director because of his extensive experience as a clinical pathologist, his substantial professional relationships with physician practices and hospital systems and his business acumen in the creation and operation of a successful pathology laboratory that developed CyPath® Lung as an LDT.
Gary Rubin — Director
Mr. Rubin, a Certified Public Accountant, serves as a Managing Member of Masters Research Partners, LLC, an investment fund of hedge funds that he co-founded in October 2000. Mr. Rubin began his career with Deloitte & Touche and later served as Managing Partner at Schissel, Rubin & Lehman, a New York-based certified public accounting firm. He has been involved in the investment business, including hedge funds, private equity, and investment banking, for more than 20 years. Mr. Rubin is active in numerous charities as well as his family’s foundation and presently serves on the board of Boca Raton Regional Hospital Foundation. He also sits on the finance committee of the Levitz Jewish Community Center. He graduated with a BS cum laude from the State University of New York at Buffalo. We believe Mr. Rubin should serve as a Director because of his financial expertise and organizational skills to his position as Chairman of the Nominating and Governance Committee and to the Board.
Family Relationships
Except for the sibling relationship between Ms. Maria Zannes and Mr. Timothy Zannes as stated above, there are no family relationships among any of our executive officers or directors.
Research Collaborator in the Development of CyPath® Lung Automated Analysis Platform
The development of the Company’s automated analysis platform for flow cytometry was developed in collaboration with Dr. Madeleine Lemieux, a bioinformatician and computational biologist who worked closely with Dr. Vivienne Rebel, Executive Vice President and Chief Medical and Science Officer and our team of scientists. Dr. Lemieux has assigned all rights to her work on behalf of bioAffinity Technologies to the Company. She is compensated at an hourly rate for her services to the Company and has been awarded stock options in compensation for her work. Her biography is provided below.
Madeleine Lemieux, Ph.D. is a bioinformatics and computational biologist who received her doctorate from the Genetics Program, University of British Columbia, Vancouver, B.C., under the supervision of Dr. Connie J. Eaves at the Terry Fox Laboratory for Hematology/Oncology. She developed an in vitro assay to distinguish blood stem cells, capable of long-term production of both myeloid and lymphoid cells, from more committed hematopoietic progenitors, and then used the assay to determine how the bone-marrow-repopulating ability of these stem cells evolves in culture. Before starting her own firm, Dr. Lemieux worked for the Department of Pediatric Oncology, Dana-Farber Cancer Institute and Harvard Medical School, Boston, Massachusetts, where she developed bioinformatics pipelines to analyze data from high-throughput platforms, including next-generation sequencing for ChIP-seq and RNA-seq as well as tiling and expression microarrays, and integrated these data with other large-scale information sources, such as gene ontology and protein-protein interaction networks, to both test hypotheses and generate testable predictions. During her time at Harvard, she also provided guidance in studies involving hematopoietic cells and, more generally, in experimental design and analysis to post-doctoral fellows and mentored two junior bioinformatics post-docs.
Science and Medical Advisory Board
The members of bioAffinity Technologies, Inc.’s Science and Medical Advisory Board (the “SMAB”) are leaders in the field of lung cancer diagnostics and flow cytometry. The SMAB provides new insights and advice to solve business problems and explore new opportunities by stimulating robust, high-quality conversations and exchange of information. The SMAB provides current knowledge, critical thinking and analysis to increase the confidence of Management in its decision-making. SMAB members have executed agreements whose provisions assure their independence and provide compensation of $2,500 per meeting for their time in attending meetings. SMAB members include:
Neil Alexis, Ph.D., Principal Investigator at the University of North Carolina School of Medicine Center for Environmental Medicine, Asthma and Lung Biology. Dr. Alexis focuses on the use of sputum as a primary sampling tool for measuring cellular, biochemical, and genetic outcomes in the human airway. Dr. Alexis is a leading expert in the use of flow cytometry in the analysis of sputum.
Sheila Habib, M.D., Director of Pulmonary Lung Nodule Clinic and the Lung Cancer Screening Program at the South Texas Veterans Health Care Systems’ Audie L. Murphy Memorial Veterans Hospital and Assistant Professor at the University of Texas Health Science Center at San Antonio. Dr. Habib specializes in pulmonary nodule management and lung cancer. Dr. Habib completed her undergraduate studies at Cornell University with a degree in Economics in 2006. She received her Doctor of Medicine from Texas Tech University School of Medicine and her residency in internal medicine and fellowship in pulmonary/critical care medicine at the University of Texas Health San Antonio.
David G. Hill, M.D., Chairman-elect of the Americna Lung Association’s National Board of Directors. Dr. Hill is a practicing pulmonary and critical care physician with Waterbury Pulmonary Associates and serves as their director of clinical research. He is an assistant clinical professor of medicine at the Yale University School of Medicine, an assistant clinical professor at the Frank Netter School of Medicine at Quinnipiac University, and a clinical instructor at the University of Connecticut School of Medicine. He has been the principal investigator for more than 75 pulmonary research trials and the author of many papers.
Catherine Sears, M.D., Assistant Professor of Medicine at Indiana University School of Medicine. Dr. Sears is a physician scientist whose laboratory focuses on the impact of DNA damage and repair on the development of smoking-related lung cancers and on treatment response. She co-chairs the pulmonary oncology and lung cancer screening programs at the Indianapolis VA Medical Center and her clinical and research interests focus on improving lung cancer screening and early lung cancer detection and treatment.
Gerard Silvestri, M.D., M.S., FCCP, Professor of Medicine and Lung Cancer Pulmonologist at the Medical University of South Carolina. Dr. Silvestri specializes in the evaluation, management, and improvement of outcomes in lung cancer patients. He has experience in evaluating new technologies for the diagnosis and staging of lung cancer. His research includes lung cancer screening, diagnosis, and staging.
| 85 |
Board of Directors Composition
Our business and affairs are managed under the direction of our Board.
Current Board of Directors
Our A&R Bylaws provide that our Board shall consist of not less than five and not more than eight directors, with such number of directors fixed by the Board. Currently, our Board consists of seven directors. Pursuant to our Charter and our A&R Bylaws, Robert A. Anderson, Stuart Diamond, Steven Girgenti, Peter S. Knight, Mohsin Y. Meghji, Gary Rubin, and Maria Zannes have been designated to serve as members of our Board. Mr. Rubin served on our Board as the Series A Representative elected separately by the holders of our Series A Preferred Stock pursuant to the Series A Director Designation Right. Following the automatic conversion of the Series A Preferred Stock shares into Common Stock immediately prior to the closing of the initial public offering, the Series A Director Designation Right ceased to exist because no shares of Series A Preferred Stock shares were outstanding. Mr. Rubin, however, will continue to serve as a director until his earlier resignation or removal or until his successor is duly elected and qualified. The number of Board seats for election by the holders of the Common Stock was expanded by one so that the director position that the holders of the Series A Preferred Stock were previously entitled to elect will be subject to election by the holders of the Common Stock following the conversion of the Series A Preferred Stock into Common Stock.
Each director on our current Board will continue to serve until such director’s successor is duly elected and qualified, or until such director’s earlier death, resignation, retirement, disqualification, or removal from office.
Committees of the Board
Our Board has established an audit committee, a compensation committee, and a nominating and corporate governance committee, each operating pursuant to a charter adopted by our Board and having the composition and responsibilities described below. The composition and functioning of all of our committees comply with all applicable requirements of the Sarbanes-Oxley Act of 2002 and with the rules and regulations of Nasdaq and the SEC. In addition, from time to time, other committees may be established under the direction of our Board to facilitate the management of our business or when necessary to address specific issues.
The members of each of our committees will serve on such committees for such term or terms as the Board may determine or until their earlier removal, resignation, or death. At least annually, each committee must review its charter and recommend any proposed changes to the Board for approval. Each committee must conduct an annual evaluation of its performance of the duties described in the committee’s charter and must present the results of the evaluation to the Board.
Audit Committee
Our audit committee consists of Stuart Diamond (Chairman), Mohsin Meghji and Gary Rubin. Our Board has determined that all members of our audit committee are independent in accordance with the requirements of Rule 10A-3 of the Exchange Act and the rules of the Nasdaq Capital Market. Our Board has also determined that Stuart Diamond and Gary Rubin are “audit committee financial experts” as defined in Item 407(d)(5)(ii) of Regulation S-K. All members of our audit committee are financially literate, as determined by our Board, and can read and understand fundamental financial statements, including the Company’s balance sheet, income statement, and cash flow statement.
Our audit committee is primarily responsible for overseeing our financial reporting and disclosure process. Among other matters, our audit committee has the following responsibilities:
| ● | selecting, retaining, compensating, overseeing, and determining the retention of an independent registered public accounting firm to audit the Company’s annual financial statements, books, records, accounts, and internal controls over financial reporting and any other registered public accounting firm engaged to prepare or issue an audit report or to perform other audit, review, or attest services for the Company; |
| ● | approving all audit engagement fees and terms and pre-approving all audit and permitted non-audit and tax services that the Company’s independent auditors or other registered public accounting firms may provide; |
| ● | establishing policies and procedures for pre-approving permitted services to be completed by the Company’s independent auditors or other registered public accounting firms on an ongoing basis; |
| ● | reviewing and discussing the results of a report prepared by the Company’s independent auditors concerning the accounting firm’s internal quality-control procedures; any material issues raised by the most recent internal quality-control review, peer review, or review by the Public Company Accounting Oversight Board (the “PCAOB”); and all relationships between the firm and the Company or any of its subsidiaries; |
| ● | reviewing and discussing with the Company’s independent auditors and management the Company’s annual audited financial statements; the adequacy and effectiveness of the Company’s internal controls; and any other matters required to be discussed by the applicable requirements of the SEC the PCAOB; |
| ● | evaluating the qualifications, performance, and independence of the Company’s independent auditors and assuring the regulator rotation of the lead audit partner; |
| ● | reviewing and discussing with the Company’s independent auditors the responsibilities of the auditors under generally accepted auditing standards; the overall audit strategy; the scope and timing of the annual audit; any significant risks identified during the auditors’ risk-assessment procedures; and the results of the annual audit; |
| ● | reviewing and discussing with the Company’s independent auditors all critical accounting policies and practices to be used in the audit; all alternative treatments of financial information within generally accepted accounting principles; and other material written communications between the auditors and management; |
| ● | reviewing and discussing with the Company’s independent auditors and management any major issues regarding accounting principles and financial-statement presentation; |
| ● | reviewing, approving, and overseeing any transaction between the Company and any related person (as defined in Item 404 of Regulation S-K) on an ongoing basis; |
| ● | informing the Company’s independent auditors of the Company’s significant relationships and transactions with related parties and reviewing and discussing with the Company’s independent auditors the auditors’ evaluation of the Company’s identification of, accounting for, and disclosure of its related-party relationships and transactions; |
| ● | recommending to the Board that the audited financial statements be included in the Company’s annual report on Form 10-K and producing the audit committee report required to be included in the Company’s proxy statement; |
| 86 |
| ● | setting the Company’s hiring policies for employees or former employees of the Company’s independent auditors that participated in any capacity in any Company audit; |
| ● | establishing and overseeing procedures for the receipt, retention, and treatment of complaints received by the Company regarding accounting, internal accounting controls, or auditing matters; |
| ● | monitoring the Company’s compliance with, investigating any alleged breach of, and enforcing the Company’s Code of Business Conduct and Ethics; |
| ● | reviewing with the Company’s General Counsel and outside legal counsel any legal and regulatory matters that could impact the Company’s financial statements; |
| ● | retaining and obtaining the advice and assistance of independent outside counsel and such other advisors as the audit committee deems necessary to fulfill its duties and responsibilities; and |
| ● | reporting regularly to the Board on the audit committee’s discussion and actions, including any significant issues or concerns that arise at the audit committee meetings. |
Compensation Committee
Our compensation committee consists of Peter Knight (Chairman), Stuart Diamond and Robert Anderson. Our Board has determined that each member of our compensation committee is independent in accordance with the rules of the Nasdaq Capital Market and the Company’s independence guidelines. Our compensation committee carries out the responsibilities delegated by the Board relating to the review and determination of executive compensation. In addition to other matters, our compensation committee is responsible for:
| ● | reviewing and approving annually the corporate goals and objectives applicable to the compensation of the chief executive officer (the “CEO”); evaluating the CEO’s performance in light of those goals and objectives; and determining and approving the CEO’s compensation level based on the compensation committee’s evaluation; | |
| ● | reviewing and approving the compensation of all of the Company’s other executive officers; | |
| ● | reviewing, making recommendations to the Board regarding, and administering the Company’s incentive-compensation plans and equity-based plans, including designating the recipients, amounts, and terms and conditions applicable to the awards to be granted under each plan; | |
| ● | reviewing and discussing with management the Company’s Compensation Discussion and Analysis (the “CD&A”); recommending that the CD&A be included in the Company’s annual report on Form 10-K and proxy statement; and producing the compensation committee report on executive-officer compensation required to be included in the Company’s annual report on Form 10-K and proxy statement; | |
| ● | reviewing the Company’s incentive-compensation arrangements to assess whether they encourage excessive risk-taking and evaluating compensation policies and practices that could mitigate any such risk; | |
| ● | reviewing and discussing at least annually the relationship between compensation and risk-management policies and practices; | |
| ● | reviewing at least annually all director compensation and benefits for service on the Board and Board committees and recommending any changes to the Board as necessary; | |
| ● | selecting, retaining, and obtaining the advice of a compensation consultant, outside legal counsel, and any other advisors as deemed necessary by the compensation committee to assist with the compensation committee’s execution of its duties and responsibilities as set forth in its charter; and | |
| ● | reporting regularly to the Board regarding the compensation committee’s actions and making recommendations to the Board as appropriate. |
Nominating and Corporate Governance Committee
Our nominating and corporate governance committee consists of Gary Rubin (Chairman), Peter Knight and Robert Anderson. Our Board has determined that each member of our nominating and corporate governance committee is independent in accordance with the rules of the Nasdaq Capital Market. Our nominating and corporate governance committee functions to carry out the responsibilities delegated by the Board relating to the Company’s director-nominations process and the development and maintenance of the Company’s corporate-governance policies. Among other matters, the responsibilities of our nominating and corporate governance committee include:
| ● | identifying and screening individuals qualified to become members of the Board, consistent with Board-approved criteria; | |
| ● | making recommendations to the Board concerning the selection and approval of director-nominees to be submitted to a stockholder vote at the annual meeting of stockholders, subject to the Board’s approval; | |
| ● | identifying and making recommendations to the Board regarding the selection and approval of candidates to fill any vacancy on the Board or any Board committee either by the stockholders’ election or the Board’s appointment; | |
| ● | developing and recommending to the Board for approval standards for determining whether a director has a relationship with the Company that would impair the director’s independence; |
| 87 |
| ● | selecting, retaining, and obtaining the advice of a director-search firm, outside counsel, and any other advisors deemed necessary to assist with the nominating and corporate governance committee’s execution of its duties and responsibilities as set forth in its charter; and | |
| ● | reporting regularly to the Board regarding the nominating and corporate governance committee’s actions and making recommendations to the Board as appropriate. |
Code of Ethics and Business Conduct
We have adopted a code of conduct applicable to our principal executive, financial and accounting officers and all persons performing similar functions. Upon the effectiveness of the registration statement of which this prospectus forms a part, our code of conduct will be available on our principal corporate website at www.bioaffinitytech.com. Information contained on our website or connected thereto does not constitute a part of, and is not incorporated by reference into, this prospectus or the registration statement of which it forms a part.
Limitations on Liability and Indemnification of Officers and Directors
Our Charter contains provisions that limit the liability of our directors for monetary damages to the fullest extent permitted by the DGCL. Consequently, our directors will not be personally liable to us or our stockholders for monetary damages for any breach of fiduciary duties as directors, except liability for:
| ● | any breach of the director’s duty of loyalty to us or our stockholders; | |
| ● | any act or omission not in good faith or that involves intentional misconduct or a knowing violation of law; | |
| ● | unlawful payments of dividends or unlawful stock repurchases or redemptions as provided in Section 174 of the DGCL; or | |
| ● | any transaction from which the director derived an improper personal benefit. |
Our Charter and our A&R Bylaws require us to indemnify our directors and officers, and allow us to indemnify other employees and agents, to the fullest extent permitted by the DGCL. Subject to certain limitations and limited exceptions, our Charter and A&R Bylaws also require us to advance expenses incurred by our directors and officers for the defense of any action for which indemnification is required or permitted.
We believe that including the limitation of liability and indemnification provisions in our Charter, A&R Bylaws, and indemnification agreements is necessary to attract and retain qualified persons such as directors, officers and key employees. Those provisions may discourage stockholders from bringing a lawsuit against our directors and officers for breaches of their fiduciary duties. They may also reduce the likelihood of derivative litigation against our directors and officers, even though an action, if successful, might benefit us and other stockholders. Further, a stockholder’s investment may be adversely affected to the extent that we pay the costs of settlement and damage awards against directors and officers as required by these indemnification provisions.
Role of the Board in Risk Oversight
One of the key functions of our Board is informed oversight of our risk management process. We face a number of risks, including those described under the section titled “Risk Factors” included in this prospectus. Our Board plays an active role in overseeing and managing our risks. The Board does not have a standing risk management committee, but rather administers this oversight function directly through the Board as a whole, as well as through its standing committees that address risks inherent in their respective areas of oversight. In particular, our Board is responsible for monitoring and assessing strategic risk exposure. Our audit committee has the responsibility to consider and discuss our major financial risk exposures and the steps our management has taken to monitor and control these exposures, including guidelines and policies to govern the process by which risk assessment and management is undertaken. The audit committee also monitors compliance with legal and regulatory requirements, in addition to oversight of the performance of our external audit function. Our nominating and corporate governance committee monitors the effectiveness of our corporate governance guidelines. Our compensation committee assesses and monitors whether any of our compensation policies and programs has the potential to encourage excessive risk-taking. While each committee will be responsible for evaluating certain risks and overseeing the management of such risks, our full Board will be regularly informed of such risks through committee reports and otherwise. While the Board oversees our risk management, management is responsible for day-to-day risk management processes. We believe this division of responsibilities enables us to address our risks most effectively.
| 88 |
EXECUTIVE COMPENSATION
We are currently considered an “emerging growth company,” within the meaning of the Securities Act, for purposes of the SEC’s executive compensation disclosure rules. In accordance with such rules, we are required to provide a Summary Compensation Table, as well as limited narrative disclosures regarding executive compensation for our last two completed fiscal years and an Outstanding Equity Awards at Fiscal Year End Table. Further, our reporting obligations extend only to our “named executive officers” (“NEOs”), meaning our principal executive officer and our next two most highly compensated executive officers in respect of their service to our Company at the end of the last completed fiscal year. Accordingly, our NEOs are:
● Maria Zannes, J.D.: President and Chief Executive Officer
● Vivienne I. Rebel, M.D., Ph.D., Executive Vice President and Chief Science and Medical Officer
● Michael Edwards, Former Chief Financial Officer (Consultant)
● Steven Girgenti, Executive Chairman
The following table sets out the compensation for our NEOs for the years ended December 31, 2022 and 2021:
| Name and Principal Position | Year |
Salary ($) |
Bonus ($) |
Restricted Stock Awards ($) (1) |
Option Awards ($) (2) |
Non-Qualified Deferred Compensation ($) |
All Other Compensation ($) (3) |
Total ($) |
||||||||||||||||||||||||
| Maria Zannes | 2022 | 253,343 | (4) | 0 | 47,944 | 0 | 0 | 7,465 | 308,752 | |||||||||||||||||||||||
| President and CEO | 2021 | 220,005 | 0 | 12,000 | 25,003 | 0 | 7,196 | 264,404 | ||||||||||||||||||||||||
| Vivienne Rebel | 2022 | 225,000 | 0 | 19,176 | 0 | 0 | 13,390 | 257,566 | ||||||||||||||||||||||||
| EVP; Chief Science & Medical Officer | 2021 | 225,000 | 0 | 13,200 | 0 | 0 | 11,590 | 249,790 | ||||||||||||||||||||||||
| Steven Girgenti | 2022 | 93,333 | (4) | 0 | 60,000 | 0 | 0 | 0 | 153,333 | |||||||||||||||||||||||
| Executive Chairman | 2021 | 60,000 | (5) | 30,000 | (6) | 90,000 | (7) | 13,322 | 90,000 | 0 | 193,222 | |||||||||||||||||||||
| Michael Edwards | 2022 | 178,579 | 0 | 66,000 | 0 | 0 | 0 | 244,579 | ||||||||||||||||||||||||
| Former Chief Financial Officer (Consultant) | 2021 | 126,866 | 0 | 0 | 5,157 | 0 | 0 | 132,023 | ||||||||||||||||||||||||
| (1) | Amounts do not reflect compensation actually received by the NEO. Instead, the amounts represent aggregate grant date fair value of the restricted stock awarded computed in accordance with ASC 718, Stock Compensation. The valuation assumptions used in determining such amounts are consistent with those described in Note 11 of the Company’s Consolidated Financial Statements for the years ended December 31, 2022, which are included in this prospectus. |
| (2) | Amounts do not reflect compensation actually received by the NEO. Instead, the amounts represent aggregate grant date fair value of options computed in accordance with ASC 718, Stock Compensation. The valuation assumptions used in determining such amounts are consistent with those described in Note 11 of the Company’s Consolidated Financial Statements for the years ended December 31, 2022, which are included in this prospectus. |
| (3) | Amounts reported as “All Other Compensation” each year for Ms. Zannes and Dr. Rebel include health benefits and premiums. |
| (4) | Includes $33,333 of fees earned or paid in cash to the NEO for his service to the Company as a director. |
| (5) | Pursuant to the terms of Mr. Girgenti’s employment agreement, Mr. Girgenti’s base salary is $120,000/year and is paid one-half in cash and one-half in the form of a grant of restricted stock. The amount reported here is the cash portion of Mr. Girgenti’s base salary. |
| (6) | Pursuant to the terms of Mr. Girgenti’s employment agreement, Mr. Girgenti’s bonus is paid one-half in cash and one-half in the form of a grant of restricted stock. Mr. Girgenti was awarded a bonus of $60,000 in each of 2020 and 2021, and the amount reported here is only the cash portion of Mr. Girgenti’s bonus. |
| (7) | Pursuant to the terms of Mr. Girgenti’s employment agreement, Mr. Girgenti’s base salary and bonus are paid one-half in cash and one-half in the form of a grant of restricted stock. The amount reported here reflects the stock portion of Mr. Girgenti’s base salary ($60,000) and the stock portion of his bonus ($30,000). |
Narrative Disclosure to Summary Compensation Table
Base Salaries
We use base salaries to recognize the experience, skills, knowledge, and responsibilities required of all our employees, including the NEOs. Base salaries are reviewed annually and adjusted from time to time to realign salaries with market levels after taking into account individual responsibilities, performance, and experience. For 2021 and 2022, the annual base salaries of our NEOs, except the salary of our former Chief Financial Officer, whose salary was based on an hourly rate, were:
| ● | For Ms. Zannes: $220,005 in 2021 and $253,343 in 2022 (which, in 2022, includes $33,333 in fees paid to Ms. Zannes for her service as a director); | |
| ● | For Dr. Rebel: $225,000 in both 2021 and 2022; | |
| ● |
For Michael Edwards: $200/hour in both 2021 and 2022 (and commencing October 20, 2021, $75.00/hour for services provided by his employees or subcontractors); and |
|
| ● | For Mr. Girgenti: $120,000 in 2021 and $153,333 in 2022 (which, in 2022, includes $33,333 in fees paid to Mr. Girgenti for his service as a director). |
Retirement Plans
We established a defined contribution plan for all employees age 21 and older who have completed one month of service for payrolls after April 1, 2022. We do not currently make a matching contribution.
Employee Benefits
Our NEOs are eligible to participate in employee benefit plans and programs, including medical and dental benefit plans.
| 89 |
Employment Agreements
The following discussion contains a summary of the terms of the Named Executive Officer employment agreements currently in effect.
Zannes Employment Agreement
We entered into an employment agreement with Ms. Zannes on February 1, 2015, which sets forth the terms and conditions of her employment (the “Zannes Agreement”). Pursuant to the Zannes Agreement, Ms. Zannes serves as our Chief Executive Officer and is entitled to an annual base salary of $220,000. The Zannes Agreement may be terminated by either party at any time, provided that Ms. Zannes is required to give us at least 90 days’ advance notice of termination.
In the event we terminate Ms. Zannes’ employment without “Cause” (as defined in the Zannes Agreement) she is entitled to receive the following payments and benefits, in addition to any accrued obligations: (i) an amount of cash equal to the sum of 12 months of her then-current annual base salary, payable in the form of salary continuation in regular installments, in accordance with our normal payroll practices, over a period of 12 months from the termination date, and (ii) reimbursement for her healthcare insurance premiums for a period of up to 12 months.
Rebel Employment Agreement
We entered into an employment agreement with Dr. Rebel on April 4, 2016, which sets forth the terms and conditions of her employment (the “Rebel Agreement”). Pursuant to the Rebel Agreement, Dr. Rebel serves as our Executive Vice President of Research and Development and Chief Medical and Science Officer and is entitled to an annual base salary of $225,000 currently. The Rebel Agreement may be terminated by either party at any time, provided that Dr. Rebel is required to give us at least 90 days’ advance notice of termination.
In the event we terminate Dr. Rebel’s employment without “Cause” (as defined in the Rebel Agreement) she is entitled to receive the following payments and benefits, in addition to any accrued obligations: (i) an amount of cash equal to the sum of 12 months of her then-current annual base salary, payable in the form of salary continuation in regular installments, in accordance with our normal payroll practices, over a period of 12 months from the termination date, and (ii) reimbursement for her healthcare insurance premiums for a period of up to 12 months.
Girgenti Employment Agreement
We entered into an employment agreement with Mr. Girgenti on January 1, 2020, which sets forth the terms and conditions of his employment (the “Girgenti Agreement”). Pursuant to the Girgenti Agreement, Mr. Girgenti serves as our Executive Chairman and is entitled to an annual base salary of $120,000, one-half of which is paid in cash and one-half of which is paid in the form of restricted stock grants. In addition, Mr. Girgenti has been awarded a bonus in 2019 and 2020 in the amount of $60,000 of which one-half is paid in cash and one-half is paid in the form of restricted stock grants. The cash portion of his compensation and bonus is deferred and credited to an unfunded bookkeeping account established on his behalf and is payable to Mr. Girgenti on the earlier of: (i) a Change in Control of the Company (as defined in the Girgenti Agreement); (ii) his termination as Chairman of the Board; (iii) the termination of his employment without Cause (as defined in the Girgenti Agreement); (iv) his death; or (v) the third anniversary of the payroll date when such compensation would have been paid but for the deferral. The Girgenti Agreement may be terminated by either party at any time, provided that Mr. Girgenti is required to give us at least 30 days’ advance notice of termination.
In the event we terminate Mr. Girgenti’s employment without “Cause” or Mr. Girgenti terminates his employment for “Good Reason” (as defined in the Girgenti Agreement) he is entitled to receive the following payments and benefits, in addition to any accrued obligations: (i) all deferred payments of his cash compensation, and (ii) the immediate vesting of any unvested shares of restricted stock granted to him under the Girgenti Agreement. In the event we terminate Mr. Girgenti’s employment for “Cause,” Mr. Girgenti will not be entitled to any of his deferred cash compensation or vesting of his restricted stock.
Consulting Agreement
Edwards Consulting Agreement
We entered into a consulting agreement with Mr. Edwards d/b/a J. Michael Edwards, LLC on May 25, 2017, as amended (the “Edwards Consulting Agreement”), pursuant to which he served as our Chief Financial Officer until his resignation on May 1, 2023. Pursuant to the Edwards Consulting Agreement, as amended on October 20, 2021, Mr. Edwards was paid at the rate of $200.00 per hour for services provided directly by him and $75.00 per hour for services provided by his employees or subcontractors. The Edwards Consulting Agreement was terminable upon notice of default or material breach, at any time upon 30 days’ written notice to the other party, or automatically upon bankruptcy, insolvency of either party or death or disability of Mr. Edwards. Upon termination of the Edwards Consulting Agreement, any unvested shares held by Mr. Edwards would be forfeited and cancelled as of the date of termination.
Executive Officers that are Not Named Executive Officers
We entered into an offer letter dated April 11, 2023, with Mr. Dougherty to serve as our Vice President and Chief Financial Officer. Pursuant to the terms of the offer letter, Mr. Dougherty receives an annual base salary of $250,000. Mr. Dougherty also received a one-time signing bonus, comprised of both cash and equity. The cash portion of the signing bonus was $30,000 and the equity portion of the signing bonus was a grant of a restricted stock award of 52,356 shares of Common Stock which was a number of shares of Company common stock equal to the quotient obtained by dividing $100,000 by the average of the closing stock price of our common stock on each of the 30 trading days prior to the date of grant. In addition, Mr. Dougherty will be eligible to receive further equity grants under our equity incentive plan at the discretion of our compensation committee and to participate in our health insurance and the 401K retirement plans on the same basis and at the same rates as the Company’s similarly situated employees.
| 90 |
Outstanding Equity Awards at December 31, 2022
The table below summarizes the outstanding equity awards awarded to our NEOs during the fiscal year ended December 31, 2022.
| Option Awards | Stock Awards | |||||||||||||||||||||||
| Name | Number of Securities Underlying Unexercised Options (#) Exercisable | Number of Securities Underlying Unexercised Options (#) Unexercisable | Option Exercise Price ($) | Option Expiration Date | Number
of Shares or Units of Stock That Have Not Vested (#) |
Market
Value of Shares of Units of Stock That Have Not Vested ($) |
||||||||||||||||||
| Maria Zannes | 64,848 | - | 1.16 | 4/28/2024 | - | - | ||||||||||||||||||
| 3,571 | - | 4.20 | 7/27/2025 | - | - | |||||||||||||||||||
| 3,571 | - | 7.00 | 7/25/2026 | - | - | |||||||||||||||||||
| 3,571 | - | 7.00 | 4/24/2027 | - | - | |||||||||||||||||||
| 7,142 | - | 7.70 | 5/7/2028 | - | - | |||||||||||||||||||
| 2,857 | - | 7.70 | 2/25/2029 | - | - | |||||||||||||||||||
| 7,142 | - | 7.70 | 7/29/2029 | - | - | |||||||||||||||||||
| 7,142 | - | 7.70 | 2/5/2030 | - | - | |||||||||||||||||||
| 7,142 | - | 7.70 | 7/27/2030 | |||||||||||||||||||||
| 7,142 | - | 7.70 | 7/26/2031 | - | - | |||||||||||||||||||
| 7,142 | - | 4.20 | 12/16/2031 | - | - | |||||||||||||||||||
| - | - | - | - | 1,888 | 3,021 | |||||||||||||||||||
| - | - | - | - | 16,591 | 26,546 | |||||||||||||||||||
| Vivienne I. Rebel | 2,857 | 7.00 | 7/25/2026 | |||||||||||||||||||||
| 2,857 | 7.70 | 11/20/2027 | ||||||||||||||||||||||
| 4,285 | 7.70 | 2/25/2029 | ||||||||||||||||||||||
| 4,285 | 7.70 | 2/5/2030 | ||||||||||||||||||||||
| - | - | - | - | 6,636 | 10,618 | |||||||||||||||||||
| Steven Girgenti | 64,848 | - | 1.16 | 4/28/2024 | ||||||||||||||||||||
| 3,571 | - | 4.20 | 7/27/2025 | |||||||||||||||||||||
| 3,571 | - | 7.00 | 7/25/2026 | |||||||||||||||||||||
| 3,571 | - | 7.00 | 4/24/2027 | |||||||||||||||||||||
| 7,142 | - | 7.70 | 5/7/2028 | |||||||||||||||||||||
| 7,142 | - | 7.70 | 7/29/2029 | |||||||||||||||||||||
| 7,142 | - | 7.70 | 7/27/2030 | |||||||||||||||||||||
| 7,142 | - | 4.20 | 12/16/2031 | |||||||||||||||||||||
| - | - | - | - | 3,896 | 6,234 | |||||||||||||||||||
| - | - | - | - | 7,792 | 12,467 | |||||||||||||||||||
| - | - | - | - | 3,896 | 6,234 | |||||||||||||||||||
| - | - | - | - | 14,285 | 22,856 | |||||||||||||||||||
| Michael Edwards | 2,857 | - | 7.70 | 02/25/2029 | - | - | ||||||||||||||||||
| 2,857 | - | 7.70 | 02/05/2030 | - | - | |||||||||||||||||||
| 2,857 | - | 7.70 | 07/26/2031 | - | - | |||||||||||||||||||
| - | - | - | - | 3,619 | 5,309 | |||||||||||||||||||
DIRECTOR COMPENSATION
The Board adopted a compensation plan for directors on November 21, 2022, pursuant to which directors are compensated for services as our director. Pursuant to this plan, for the period beginning September 6, 2022, through December 31, 2022, each director received a single, lump-sum cash payment of $33,333. Beginning on January 1, 2023, each director will receive compensation in accordance with the following:
| ● | each director will be paid $25,000 per year in cash with payments to be made in four quarterly installments of $6,250 on each of January 1, April 1, July 1, and October 1; | |
| ● | the Chairman of the Board will be paid an additional $10,000 per year in cash, with payments to be made to the chairman in four quarterly installments of $2,500 on each of January 1, April 1, July 1, and October 1; | |
| ● | the chairman of the Audit Committee will be paid an additional $5,000 per year in cash, with payments to be made to the Audit Committee chairman in four quarterly installments of $1,250 on each of January 1, April 1, July 1, and October 1; | |
| ● | the chairmen of the Compensation Committee and the Nominating and Governance Committee will be paid an additional $2,500 per year in cash, with payments to be made to such committee chairmen in four quarterly installments of $625 on each of January 1, April 1, July 1, and October 1; and |
| 91 |
| ● | each director will be granted a quarterly restricted stock award valued at $18,750 based on the fair market value of the Company’s Common Stock on the date of grant (which shall be deemed to be the greater of (a) the average closing price of the Company’s Common Stock on the Nasdaq Stock Market over the 30 trading days prior to the date of grant, or (b) the closing price of the Company’s Common Stock on the Nasdaq Stock Market on the trading date immediately prior to the date of grant) on each of January 1, April 1, July 1, and October 1. The restricted stock awards will vest after three months of continued service by the director. |
The table below summarizes the compensation paid to our non-employee directors for the fiscal year ended December 31, 2022.
| Name |
Fees Earned or Paid in Cash ($) |
Option Awards ($) (1) |
Total ($) |
|||||||||
| Robert Anderson | 33,333 | 33,333 | ||||||||||
| Stuart Diamond | 33,333 | 20,278 | 53,611 | |||||||||
| Peter Knight | 33,333 | 33,333 | ||||||||||
| Mohsin Meghji | 33,333 | 33,333 | ||||||||||
| Gary Rubin | 33,333 | 33,333 | ||||||||||
| (1) | Amounts do not reflect compensation actually received by the non-employee director. Instead, the amounts represent aggregate grant date fair value of options computed in accordance in accordance with ASC 718, Stock Compensation. The valuation assumptions used in determining such amounts are consistent with those described in Note 11 of the Company’s Consolidated Financial Statements for the years ended December 31, 2022, which are included in prospectus. Stuart Diamond was granted 7,142 stock options on March 17, 2022. As of December 31, 2022, 5,357 options were vested. |
The table below shows the aggregate number of option awards outstanding at fiscal year-end of our non-employee directors.
| Name |
Number of Shares Subject to Outstanding Options
as of December 31, 2022 |
|||
| Robert Anderson | 104,129 | |||
| Stuart Diamond | 7,142 | |||
| Peter Knight | 28,568 | |||
| Mohsin Meghji | 21,426 | |||
| Gary Rubin | 32,139 | |||
Equity Compensation Plan Information
The following table sets forth information as of December 31, 2022, with respect to our 2014 Equity Incentive Plan (the “Plan”), which was approved by the Company’s stockholders:
| Plan Category |
Number of securities to be issued upon exercise of outstanding options, warrants and rights (a) |
Weighted average exercise price of outstanding options, warrants and rights (b) |
Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a)) |
|||||||||
| Equity compensation plans approved by security holders | 806,392 | $ | 4.33 | 58,116 | ||||||||
| Total | 806,392 | $ | 4.33 | 58,116 | ||||||||
At our annual stockholders meeting held on June 6, 2023, our stockholders approved an amendment to the Plan, to increase the number of awards available under the Plan to 2,000,000 awards.
| 92 |
PRINCIPAL STOCKHOLDERS
The following table sets forth information regarding the beneficial ownership of shares of our Common Stock as of September 20, 2023, by (1) each person known to us to beneficially own more than 5% of any class of the Company’s outstanding voting securities, (2) each of our directors and named executive officers, and (3) all of our current directors and executive officers as a group.
Beneficial ownership is determined according to the rules of the SEC, which generally provide that a person has beneficial ownership of a security if he, she or it possesses sole or shared voting or investment power over that security, including options and warrants that are currently exercisable or exercisable within 60 days. In computing the number of shares beneficially owned by a person or entity and the percentage ownership of that person or entity in the table below, all shares subject to options and warrants were deemed outstanding if such securities are currently exercisable or will vest within 60 days of the filing of September 20, 2023. These shares were not deemed outstanding, however, for the purpose of computing the percentage ownership of any other person or entity.
The percentage of beneficial ownership of our Common Stock is based on 9,350,297 shares of Common Stock outstanding as of September 20, 2023, which includes 133,414 unvested shares of restricted Common Stock which are therefore subject to forfeiture, but which are outstanding and have voting rights, and 564,972 shares of Common Stock issued to the Joyce Trust pursuant to the terms of the Asset Purchase Agreement.
Unless otherwise indicated, we believe that each person named in the table below has sole voting and investment power with respect to all shares of Common Stock beneficially owned by such person.
| Name and Address(1) | Number of Shares of Common Stock | Percent of Common Stock Owned Before the Offering(2) | Percent of Common Stock Owned After the Offering(2) | |||||||||
| Directors and Executive Officers: | ||||||||||||
| Maria Zannes(3) | 237,508 | 2.51 | % | 1.89 |
% | |||||||
| Vivienne Rebel(4) | 28,665 | * | * | |||||||||
| Steven Girgenti(5) | 1,027,789 | 10.87 | % | 8.20 |
% | |||||||
| Michael Edwards(6) | 47,829 | * | * | |||||||||
| Timothy Zannes(7) | 80,606 | * | * | |||||||||
| Robert Anderson(8) | 156,598 | 1.65 | % | 1.25 | % | |||||||
| Stuart Diamond(9) | 59,611 | * | * | |||||||||
| Mohsin Meghji(10) | 119,296 | 1.27 | % | * | ||||||||
| Peter Knight(11) | 119,405 | 1.27 | % | * | ||||||||
| Gary Rubin(12) | 1,696,265 | 18.08 | % | 13.60 | % | |||||||
| Roby Joyce(13) | 564,972 | 6.04 |
% | 4.54 | % | |||||||
|
All Directors and Current Executive Officers as a Group (12 Individuals(14)): |
4,154,545 | 41.76 | % | 31.87 | % | |||||||
| Five Percent Holders: | ||||||||||||
| The Harvey Sandler Revocable Trust(15) | 1,584,144 | 16.94 | % | 12.74 | % | |||||||
| Nathan Perlmutter(16) | 495,683 | 5.30 | % | 3.99 | % | |||||||
| The Joyce Living Trust(17) | 564,972 |
6.04 |
% | 4.54 |
% | |||||||
| * | Ownership of less than 1%. |
| (1) | Unless otherwise indicated, the address for each person is c/o bioAffinity Technologies, Inc., 22211 West Interstate 10, Suite 1206, San Antonio, Texas, 78257. |
| (2) | Based on 9,350,297 shares of Common Stock outstanding as of September 20, 2023, which includes: (i) 133,414 unvested shares of restricted Common Stock, which are therefore subject to forfeiture, but which are outstanding and have voting rights; and (ii) 564,972 shares of Common Stock issued to the Joyce Trust pursuant to the terms of the Asset Purchase Agreement. |
| (3) | Includes (i) 39,998 shares of Common Stock owned by Ms. Zannes of record or beneficially; (ii) 76,240 shares of Common Stock issued to Ms. Zannes as restricted stock; and (iii) 121,270 shares of Common Stock issuable upon exercise of options with a weighted average exercise price of $3.64 per share granted to Ms. Zannes that are immediately exercisable. Does not include the following outstanding warrants that were amended by the Warrant Amendment and therefore are not exercisable: (i) 23,571 shares of Common Stock underlying warrants with a term of five years having an exercise price of $6.125 per share; and (ii) 79,995 shares of Common Stock underlying warrants with a term of five years having an exercise price of $3.0625 per share. Also does not include the issuance of restricted shares of Common Stock that will be issued to Ms. Zannes, as part of our director compensation policy on October 1, 2023, which number of shares will be determined by dividing $18,750 by the greater of: (i) the average of the closing stock price of our common stock on each of the 30 trading days prior to the date of grant; or (ii) the closing price of our Common Stock on the trading date immediately prior to the date of grant. |
| (4) | Includes (i) 14,381 shares of Common Stock issued to Dr. Rebel as restricted stock; and (ii) 14,284 shares of Common Stock issuable upon exercise of options with a weighted average exercise price of $7.56 per share granted to Dr. Rebel that are immediately exercisable. |
| (5) | Includes (i) 830,129 shares of Common Stock owned by Mr. Girgenti of record or beneficially; (ii) 84,576 shares of Common Stock issued to Mr. Girgenti as restricted stock; (iii) 8,955 shares of Common Stock owned directly by the Cranye Girgenti Testamentary Trust, for which Mr. Girgenti serves as trustee; and (iv) 104,129 shares of Common Stock issuable upon exercise of options with a weighted average exercise price of $3.164 per share granted to Mr. Girgenti that are immediately exercisable. Does not include the following outstanding warrants that were amended by the Warrant Amendment and therefore are not exercisable: (i) 345,252 shares of Common Stock underlying warrants with a term of five years having an exercise price of $6.125 per share; (ii) 123,811 shares of Common Stock underlying warrants with a term of five years having an exercise price of $5.25 per share; (iii) 200,484 shares of Common Stock underlying warrants with a term of five years having an exercise price of $3.0625 per share; (iv) 5,952 shares of Common Stock underlying warrants having an exercise price of $6.125 per share held by the Cranye Girgenti Testamentary Trust; and (v) 2,380 shares of Common Stock underlying warrants having an exercise price of $5.25 per share held by the Cranye Girgenti Testamentary Trust. Also does not include the issuance of restricted shares of Common Stock that will be issued to Mr. Girgenti, as part of our director compensation policy on October 1, 2023, which number of shares will be determined by dividing $18,750 by the greater of: (i) the average of the closing stock price of our common stock on each of the 30 trading days prior to the date of grant; or (ii) the closing price of our Common Stock on the trading date immediately prior to the date of grant. |
| (6) | Mr. Edwards resigned as our Chief Financial Officer on May 1, 2023. Includes (i) 30,118 shares of Common Stock owned directly by Mr. Edwards of record; (ii) 3,619 shares of Common Stock issued to Mr. Edwards as restricted stock; (iii) 1,903 shares of Common Stock underlying a warrant with a term of five years having an exercise price of $6.125 per share; (iv) 761 shares of Common Stock underlying a warrant with a term of five years having an exercise price of $5.25 per share; and (v) 11,428 shares of Common Stock issuable upon exercise of options with a weighted average exercise price of $7.70 per share granted to Mr. Edwards that are immediately exercisable. |
| 93 |
| (7) | Includes (i) 76,987 shares of Common Stock issuable upon exercise of options with a weighted average exercise price of $2.121 per share granted to Mr. Zannes that are immediately exercisable; and (ii) 3,619 shares of Common Stock issued to Mr. Zannes as restricted stock. |
| (8) | Includes (i) 4,081 shares of Common Stock owned by Mr. Anderson of record or beneficially; (ii) 28,392 shares of Common Stock issued to Mr. Anderson as restricted stock; (iii) 104,129 shares of Common Stock issuable upon exercise of options with a weighted average exercise price of $3.164 per share granted to Mr. Anderson that are immediately exercisable; and (iv) 19,996 shares of Common Stock underlying warrants with a term of five years having an exercise price of $3.0625. Does not include the issuance of restricted shares of Common Stock that will be issued to Mr. Anderson, as part of our director compensation policy on October 1, 2023, which number of shares will be determined by dividing $18,750 by the greater of: (i) the average of the closing stock price of our common stock on each of the 30 trading days prior to the date of grant; or (ii) the closing price of our Common Stock on the trading date immediately prior to the date of grant. |
| (9) | Includes (i) 4,081 shares of Common Stock owned by Mr. Diamond of record or beneficially; (ii) 28,392 shares of Common Stock issued to Mr. Diamond as restricted stock; (iii) 7,142 shares of Common Stock issuable upon exercise of options with a weighted average exercise price of $4.20 per share granted to Mr. Diamond that are immediately exercisable; and (iv) 19,996 shares of Common Stock underlying warrants with a term of five years having an exercise price of $3.0625 per share. Does not include the issuance of restricted shares of Common Stock that will be issued to Mr. Diamond, as part of our director compensation policy on October 1, 2023, which number of shares will be determined by dividing $18,750 by the greater of: (i) the average of the closing stock price of our common stock on each of the 30 trading days prior to the date of grant; or (ii) the closing price of our Common Stock on the trading date immediately prior to the date of grant. |
| (10) | Includes (i) 69,478 shares of Common Stock owned directly by Mr. Meghji of record; (ii) 28,392 shares of Common Stock issued to Mr. Meghji as restricted stock; and (iii) 21,426 shares of Common Stock issuable upon exercise of options with a weighted average exercise price of $7.308 per share granted to Mr. Meghji that are immediately exercisable. Does not include the issuance of restricted shares of Common Stock that will be issued to Mr. Meghji, as part of our director compensation policy on October 1, 2023, which number of shares will be determined by dividing $18,750 by the greater of: (i) the average of the closing stock price of our common stock on each of the 30 trading days prior to the date of grant; or (ii) the closing price of our Common Stock on the trading date immediately prior to the date of grant. |
| (11) | Includes (i) 22,448 shares of Common Stock owned directly by Mr. Knight of record; (ii) 28,392 shares of Common Stock issued to Mr. Meghji as restricted stock; (iii) 28,568 shares of Common Stock issuable upon exercise of options with a weighted average exercise price of $7.434 per share granted to Mr. Knight that are immediately exercisable; and (iv) 39,997 shares of Common Stock underlying warrants with a term of five years having an exercise price of $3.0625 per share. Does not include the issuance of restricted shares of Common Stock that will be issued to Mr. Knight, as part of our director compensation policy on October 1, 2023, which number of shares will be determined by dividing $18,750 by the greater of: (i) the average of the closing stock price of our common stock on each of the 30 trading days prior to the date of grant; or (ii) the closing price of our Common Stock on the trading date immediately prior to the date of grant. |
| (12) | Includes (i) 51,590 shares of Common Stock owned directly by Mr. Rubin of record; and (ii) 28,392 shares of Common Stock issued to Mr. Rubin as restricted stock. Does not include the following outstanding warrants that were amended by the Warrant Amendment and therefore are not exercisable: (i) 12,241 shares of Common Stock underlying warrants with a term of five years having an exercise price $6.125 per share; (ii) 4,896 shares of Common Stock underlying warrants with a term of five years having an exercise price of $5.25 per share; (iii) 32,139 shares of Common Stock issuable upon exercise of options with a weighted average exercise price of $7.469 per share granted to Mr. Rubin that are immediately exercisable; (iv) 1,584,144 shares of Common Stock owned by the Harvey Sandler Revocable Trust, for which Mr. Rubin serves as co-trustee; (v) 408,125 shares of Common Stock underlying warrants with a term of five years having an exercise price $6.125 per share held by the Harvey Sandler Revocable Trust; (vi) 163,248 shares of Common Stock underlying warrants with a term of five years having an exercise price of $5.25 per share held by the Harvey Sandler Revocable Trust; (vii) 79,995 shares of Common Stock underlying warrants with a term of five years having an exercise price of $3.0625 per share held by the Harvey Sandler Revocable Trust. Also does not include the issuance of restricted shares of Common Stock that will be issued to Mr. Rubin, as part of our director compensation policy on October 1, 2023, which number of shares will be determined by dividing $18,750 by the greater of: (i) the average of the closing stock price of our common stock on each of the 30 trading days prior to the date of grant; or (ii) the closing price of our Common Stock on the trading date immediately prior to the date of grant. |
| (13) |
Includes 564,972 shares of Common Stock owned by the Joyce Living Trust. Dr. Joyce is co-trustee of the Joyce Living Trust, together with his wife, Joyce M. Joyce, each of whom may act unilaterally with regard to voting and disposition power over the shares held by the Joyce Living Trust. The Joyce Living Trust has an address at 1092 Madeline Street, New Braunfels, Texas 78132. Does not include 14,285 shares of Common Stock underlying warrants with a term of five years having an exercise price of $5.25 per share that were amended by the Warrant Amendment and therefore are not exercisable. Also does not include the issuance of restricted shares of Common Stock that will be issued to Dr. Joyce, as part of our director compensation policy on October 1, 2023, which number of shares will be determined by dividing $18,750 by the greater of: (i) the average of the closing stock price of our common stock on each of the 30 trading days prior to the date of grant; or (ii) the closing price of our Common Stock on the trading date immediately prior to the date of grant. |
| (14) | Excludes shares of Common Stock owned by Michael Edwards. Includes shares owned by our current executive officers that are “named executive officers: including: (i) 52,356 shares of restricted Common Stock owned by Michael Dougherty, our Chief Financial Officer; and (ii) 3,619 shares of restricted Common stock owned by Xavier Reveles and 7,855 shares issuable upon exercise of options granted to Xavier Reveles that are immediately exercisable or exercisable within 60 days of September 20, 2023. |
| (15) | Includes 1,584,144 shares of Common Stock owned by the Trust either of record or beneficially . Does not include the following outstanding warrants that were amended by the Warrant Amendment and therefore are not exercisable: (i) 408,125 shares of Common Stock underlying warrants with a term of five years having an exercise price $6.125 per share; (ii) 163,248 shares of Common Stock underlying warrants with a term of five years having an exercise price of $5.25 per share; (iii) 79,995 shares of Common Stock underlying warrants with a term of five years having an exercise price of $3.0625. |
| (16) | Includes (i) 327,909 shares of Common Stock owned directly by Mr. Perlmutter of record; (ii) 119,839 shares of Common Stock underlying warrants with a term of five years having an exercise price of $6.125 per share; and (iii) 47,935 shares of Common Stock underlying warrants with a term of five years having an exercise price of $5.25. |
| (17) | Includes 564,972 shares of Common Stock owned by the Joyce Living Trust. Dr. Joyce is co-trustee of the Joyce Living Trust , together with his wife, Joyce M. Joyce, each of whom may act unilaterally with regard to voting and disposition power over the shares held by the Joyce Living Trust. The Joyce Living Trust has an address at 1092 Madeline Street, New Braunfels, Texas 78132. Does not include 14,285 shares of Common Stock underlying warrants with a term of five years having an exercise price of $5.25 per share that were amended by the Warrant Amendment and therefore are not exercisable. |
| 94 |
CERTAIN RELATIONSHIPS AND RELATED PARTY TRANSACTIONS
Related-Party Transactions
In addition to the compensation arrangements with directors and executive officers described under “Executive Compensation,” the following is a description of each transaction since January 1, 2021, and each currently proposed transaction in which:
| ● | the Company was or is to be a participant; | |
| ● | the amount involved exceeds $120,000; and | |
| ● | any related person had or will have a direct or indirect material interest. |
Zannes Purchase of Bridge Note and Accompanying Bridge Warrant
In August 2022, Maria Zannes, our President, CEO, and a director, purchased a convertible promissory note (a “Bridge Note”) in the principal amount of $99,000, which accrued interest at a rate of 6% per year. Originally, all principal and unpaid interest on the Bridge Note was due, if not settled prior, on May 31, 2022. Upon the closing of our initial public offering on September 6, 2022, the Bridge Note automatically converted into 23,672 shares of Common Stock.
In connection with her Bridge Note purchase, Ms. Zannes was issued an accompanying warrant (a “Bridge Warrant”) to purchase one share of our Common Stock for each conversion share based on the principal balance of the Bridge Note at an exercise price equal to $5.25 per share. The Bridge Warrant issued to Ms. Zannes entitled her to purchase 23,571 shares of Common Stock at an exercise price of $5.25 per share.
Edwards Note
On June 12, 2020, Michael Edwards, former CFO, purchased a convertible promissory note from us with a principal amount of $7,995 The note bore interest at 8% per annum and matures on October 31, 2022. The unpaid principal and accrued interest under the note could be converted into shares of our Common Stock at a conversion price of $4.20 per share. On July 25, 2022, Mr. Edwards agreed to extend the maturity date of his note to October 31, 2022, on the same terms provided to all other convertible noteholders in exchange for our issuance to him of a warrant to purchase 761 shares of Common Stock at an exercise price equal to $5.25 per share. The note automatically converted into 2,229 shares of Common Stock upon the completion of our initial public offering.
Perlmutter Note
On October 22, 2020, Nathan Perlmutter purchased a convertible promissory note from us with a principal amount of $100,000. The note bore interest at 8% per annum and matures on October 31, 2022. The unpaid principal and accrued interest under the note could be converted into shares of the Company’s Common Stock at a conversion price of $4.20 per share. On July 25, 2022, Mr. Perlmutter agreed to extend the maturity date of his note to October 31, 2022, on the same terms provided to all other convertible noteholders in exchange for our issuance to him of a warrant to purchase 47,935 shares of Common Stock at an exercise price equal to $5.25 per share. The note automatically converted into approximately 27,201 shares of Common Stock upon the completion of our initial public offering.
Girgenti Purchase of Bridge Note and Accompanying Bridge Warrant
On each of January 13, 2021, March 10, 2021, March 24, 2021, June 8, 2021, July 3, 2021 and August 11, 2022, Steven Girgenti, the Executive Chairman and a director of the Company, purchased a Bridge Note in the principal amount of $10,000, $450,000 $40,000, $150,000, $60,000 and $150,000, respectively. The Bridge Notes bear interest at 8% per annum and matured on October 31, 2022, except the note issued on July 3, 2021 matured on December 31, 2022. On July 25, 2022, Mr. Girgenti agreed to extend the maturity date of his note to October 31, 2022, on the same terms provided to all other convertible noteholders in exchange for our issuance to him of a warrant to purchase 47,935 shares of Common Stock at an exercise price equal to $5.25 per share. The notes together with previously issued notes automatically converted into approximately 81,516 shares of Common Stock upon the closing of our initial public offering based on the outstanding principal balance and accrued and unpaid interest under such note as of August 3, 2022 at a conversion price of $4.20 per share.
| 95 |
PPLS Acquisition of the Laboratory
On September 18, 2023, PPLS consummated the Acquisition of the Laboratory Assets pursuant to the terms of the Asset Purchase Agreement with Village Oaks. As a result of the Acquisition, the CAP-accredited CLIA-certified clinical pathology laboratory is owned by PPLS. Dr. Joyce was the Medical Director and Laboratory Director of the clinical pathology laboratory prior to the Acquisition and he continues to serve as Medical Director and Laboratory Director after the Acquisition. Founded in 2007 by Dr. Roby Joyce, Village Oaks has provided pathology services to physicians practicing in a variety of outpatient settings. Since September 2021, Village Oaks, under the trade name Precision Pathology Services, has offered CyPath® Lung for sale as an LDT for the detection of early-stage lung cancer. In addition to CyPath® Lung, PPLS intends to continue to offer a range of laboratory services including respiratory testing for SARS-CoV-2 and influenza, anatomical pathology, morphological stains, histological services, DNA extractions, STI testing and women’s and men’s health testing.
Pursuant to the terms of the Asset Purchase Agreement, PPLS acquired the Laboratory Assets, which included all of the assets owned by Village Oaks, other than medical assets, which are assets Village Oaks used in connection with its management and operation of a clinical pathology laboratory, now owned by PPLS, and related services business, and assumed certain liabilities and obligations. The laboratory is a clinical pathology laboratory that is CAP-accredited and CLIA-certified. Pursuant to the terms of the Asset Purchase Agreement, Village Oaks received $3,500,000 in consideration for the assets to be purchased by PPLS, of which $1,000,000 was paid by the issuance of 564,972 shares of our restricted Common Stock to the Joyce Trust, which share number was determined by dividing $1,000,000 by $1.77, the average of the trading day closing prices for the 30 days prior to September 15, 2023, rounded to the nearest whole share.
The Asset Purchase Agreement contains customary representations, warranties and covenants made by PPLS and Village Oaks and consummation of the transaction was subject to customary closing conditions, including, among other things, entry into the other ancillary agreements described below.
Pursuant to the Asset Purchase Agreement, PPLS assumed all liabilities and obligations under and obtained any and all rights, title and interest of Village Oaks in and to (i) the Assumed Leases under the Assumption Agreement; (ii) the Assumed Contracts under the Assumption Agreement; (iii) all accounts payable of Village Oaks as of September 18, 2023 that were incurred in the ordinary course of business consistent with past custom and practice; and (iv) the lease of the premises used in connection with operation of the CLIA-certified and CAP-accredited clinical pathology laboratory, pursuant to the Assignment and Assumption of Lease, which Assignment of Lease was consented to by the landlord of the leased premises. The monthly rent is currently $10,143.83 per month and the term of the Lease is five years.
In connection with the Asset Purchase Agreement, PPLS entered into the Management Services Agreement with Village Oaks, pursuant to which PPLS will provide comprehensive management and administrative services to Village Oaks in connection with the operation of Village Oaks’ professional cytopathology, histopathology, clinical and anatomic pathology interpretation medical services practice. PPLS will provide space, equipment, administrative, management and clinical personnel, billing and collection, and related management services to Village Oaks in exchange for a management fee of 70% of the net revenues received by Village Oaks from the provision of the medical services. The Management Services Agreement has an initial term of 20 years and provides that upon expiration of the initial term, it will be automatically extended for two additional successive terms of five years each, unless either party delivers written notice of its intention not to extend the term of the agreement not less than 90 days prior to the expiration of the preceding term. The Management Services Agreement also provides that until the fifth anniversary of its effective date, Village Oaks will not, without the prior written approval of PPLS own, operate or have any financial interest in any other person or entity that operates an independent laboratory or an enterprise within the United States that provides or promotes management or administrative services or any product or services substantially similar to those provided by PPLS.
In connection with the Asset Purchase Agreement, PPLS entered into the Succession Agreement, pursuant to which Dr. Joyce, as holder of 100% of the issued and outstanding stock of Village Oaks, and Village Oaks are restricted from disposing of their equity interests in Village Oaks, subject to certain exceptions, without the prior written consent of us and Village Oaks. The Succession Agreement further provides that the entire equity interest held by Dr. Joyce in Village Oaks will be automatically assigned and transferred to a successor who meets the Eligibility Requirements of a Designated Physician, as such terms are defined and described in the Succession Agreement, in the event of, among other things, the death, disability, retirement, or a court’s determination of incompetence of Dr. Joyce, as well as Dr. Joyce’s failure to satisfy the eligibility requirements of a Designated Physician, exclusion or disqualification from participation in the Medicare program, conviction of a felony or crime or moral turpitude, bankruptcy filing, or material breach of the Succession Agreement. In the event of the automatic transfer of Dr. Joyce’s equity interests in Village Oaks as provided in the Succession Agreement, such agreement provides that the board of directors of Village Oaks shall nominate a group of three candidates as the Designated Physician who satisfy the Eligibility Requirements. In the event the Company desires not to select any of such candidates, the Company shall select and appoint a successor Designated Physician from any other physicians that satisfy the Eligibility Requirements. Subject in all cases to the Management Services Agreement, Dr. Joyce shall not cause any voluntary interruption of the conduct of Village Oaks’ business and operations, and shall use commercially reasonable efforts to preserve (or assist us in preserving) all rights, privileges and franchises held by Village Oaks, including the maintenance of all contracts, copyrights, trademarks, licenses and registrations.
| 96 |
In connection with the Asset Purchase Agreement, PPLS entered into the Professional Services Agreement with Village Oaks, pursuant to which Village Oaks will provide pathology interpretation services as requested on behalf of PPLS based on the professional fees approved for the CPT code for the services provided under the Medicare Physician Fee Schedule in the locality where the test is performed. The Professional Services Agreement has an initial term of 20 years and provides that upon expiration of the initial term, it will be automatically extended for successive terms of 12 months each, unless either party delivers written notice of its intention not to extend the term of the agreement not less than 30 days prior to the expiration of the preceding term.
In connection with the Asset Purchase Agreement, we entered into the Executive Employment Agreement with Dr. Joyce, for a term of three years, to serve as the Medical Director and Laboratory Director of PPLS at a base salary of $333,333.34 per year. Pursuant to the Joyce Employment Agreement, Dr. Joyce was also appointed to serve on our Board of Directors. Dr. Joyce will be eligible to participate in or receive benefits under our benefit plans generally made available to executives of similar status and responsibilities and will be provided use of a company car. In the event the Joyce Employment Agreement is terminated for any reason, including by Dr. Joyce upon 60 days’ notice, by us for cause or by reason of Dr. Joyce’s death, Dr. Joyce (or his estate as applicable) will receive his base salary for the remainder of the three-year employment term. However, the Joyce Employment Agreement provides that if Dr. Joyce breaches any of the restrictive covenants set forth in the Joyce Employment Agreement, including a covenant not to compete during his term of employment and a covenant not to knowingly disclose confidential information, such breach will be grounds for the immediate termination of Dr. Joyce and will result in the forfeiture of all compensation and benefits otherwise due to Dr. Joyce.
One of the Assumed Leases is the Hologic Equipment Lease, pursuant to which PPLS leases reagent equipment from Hologic and is required to purchase a minimum number of specified testing kits each year. The total monthly minimum purchase commitment PPLS is required to pay Hologic, inclusive of the lease of the reagent equipment, is $16,914 per month. The term of the Hologic Equipment Lease currently expires on December 20, 2027.
Another of the Assumed Leases is the Leica Equipment Lease, pursuant to which PPLS leases reagent equipment from Leica and is required to purchase a minimum number of specified testing kits. The total monthly minimum purchase commitment PPLS is required to pay to Leica, inclusive of the lease of the reagent equipment, is $19,790 per month. The term of the Leica Equipment Lease currently expires on March 23, 2026.
One of the Assumed Contracts is the License Agreement. Pursuant to the License Agreement, Pathology Watch granted a license to its digital imaging cloud-based pathology platform to facilitate remote interpretation and billing of pathology specimens by qualified professionals to PPLS for a monthly fee of $25,000. In connection with the License Agreement, Pathology Watch also provides certain support services and marketing vendor services to PPLS for the monthly fee of $38,000, for a total monthly fee paid by PPLS to Precision Watch of $63,000. The License Agreement is for an initial term of 12 months, unless terminated by either party upon 90 days’ notice, and provides that upon expiration of the initial term (or any renewal term), it will be automatically extended for successive 12-month terms, unless either party notifies the other party of its intention not to renew the License Agreement not less than 90 days prior to the expiration of the current term.
In connection with the Asset Purchase Agreement, Dr. Joyce, on behalf of Village Oaks, executed the Bill of Sale, pursuant to which all rights, title, and interest of Village Oaks in and to the permits listed on Exhibit A attached thereto, inclusive of the CLIA-certificate and CAP-accreditation, notwithstanding the transfer of the CLIA certificate by operation of law to PPLS upon consummation of the Acquisition, were confirmed to have been transferred and assigned to PPLS.
Amendment to Warrants
On September 17, 2023, Mr. Girgenti, the Cranye Girgenti Testamentary Trust, Gary Rubin, The Harvey Sandler Revocable Trust, a trust of which Mr. Rubin is a co-trustee, Ms. Zannes and Dr. Joyce consented to an amendment of the terms of the outstanding warrants that they own. Such warrants include warrants (i) Tradeable Warrants to purchase 98,198, 39,182, and 39,182 shares of Common Stock owned by Mr. Girgenti, The Harvey Sandler Revocable Trust, and Ms. Zannes, respectively); (ii) Non-Tradeable Warrants to purchase 102,286, 40,813, and 40,813 shares of Common Stock owned by Mr. Girgenti, The Harvey Sandler Revocable Trust, and Ms. Zannes, respectively; and (iii) Pre-IPO Warrants to purchase 469,063, 8,332, 571,373, 23,571, 17,137, and 14,285 shares of Common Stock owned by Mr. Girgenti, the Cranye Girgenti Testamentary Trust, Mr. Rubin, The Harvey Sandler Revocable Trust, Ms. Zannes and Dr. Joyce, respectively. The warrant amendment provides that such warrants will not be exercisable until the date that we file a certificate of amendment to our certificate of incorporation with the State of Delaware which increases the number of shares of our authorized Common Stock to allow for sufficient authorized and unissued shares of Common Stock for the full exercise of all of the outstanding Pre-IPO Warrants, Tradeable Warrants and Non-Tradeable Warrants of the Company and the issuance of all of the shares of Common Stock underlying such warrants.
Policies and Procedures for Related Party Transactions
The Board has adopted a written Code of Ethics and Business Conduct, which includes a policy on conflicts of interest that requires our directors and executive officers to seek determination and prior authorizations or approvals of potential conflicts of interest exclusively from the Board. In reviewing and approving any such transactions, the Company’s General Counsel and Board consider all relevant facts and circumstances. The Code of Ethics and Business Conduct is available on the Investor Relations section of the Company’s website at ir.bioaffinitytech.com and can be accessed through the “Governance Documents” hyperlink under the “Governance” tab. We intend to disclose any amendments to the Code of Ethics and Business Conduct, or any waivers of its requirements, on its website to the extent required by the applicable rules and exchange requirements.
Insider Trading Policy
We maintain a policy on insider trading that applies to any and all transactions in our securities held by any director, officer, or employee. The policy prohibits all of our directors, officers, and employees from trading in our securities while in possession of material nonpublic information (“MNPI”) about us and from giving MNPI to others who may trade on the basis of such information. Under the policy, Timothy Zannes, the Company’s Executive Vice President, Secretary, and General Counsel, is designated as our Insider Trading Compliance Officer (the “Compliance Officer”). Prior to engaging in transfers of our securities intended to comply with the affirmative defense provided under Rule 10b5-1 promulgated under the Exchange Act, employees, officers, and directors must receive the Compliance Officer’s approval.
Transfer Agent
The transfer agent and registrar for the Company’s Common Stock is VStock Transfer, LLC. The transfer agent and registrar’s address is 18 Lafayette Place, Woodmere, New York 11598.
| 97 |
DESCRIPTION OF SECURITIES
We are offering Units in this Offering at an assumed Offering Price of $1.62. Each Unit consists of one share of our Common Stock and one five-year Warrant to purchase one share of our Common Stock at an exercise price equal to $[●], which is 120% of the Offering Price per Unit. The Units will not be certificated or issued in stand-alone form. The shares of our Common Stock and the Warrants underlying the Units are immediately separable upon issuance and will be issued separately in this Offering. We are also registering the shares of Common Stock issuable upon exercise of the Warrants. These securities are being issued pursuant to an underwriting agreement between us and the underwriters. You should review the underwriting agreement, the form of Warrant filed as an exhibit to the registration statement of which this prospectus is a part, for a complete description of the terms and conditions applicable to the Warrants.
The following summary describes the material terms of our capital stock and provisions of our Charter and our A&R Bylaws. This summary does not purport to be complete and is qualified in its entirety by reference to all of the provisions of our Charter and our A&R Bylaws, which are filed as exhibits to the registration statement of which this prospectus is a part.
Our Board of Directors and stockholders approved an amendment to our Charter to effect a 1-for-7 reverse stock split of our Common Stock in connection with our initial public offering. As a result of the reverse stock split every seven shares of our outstanding Common Stock was combined and reclassified into one share of our Common Stock. No fractional shares were issued in connection with the reverse stock split, and any of our stockholders that were entitled to receive a fractional share as a result of the reverse stock split instead received cash in lieu of a fractional share valued at the per-Unit price of the initial public offering. The reverse stock split became effective on June 23, 2022. The reverse stock split was intended to allow us to meet the minimum share price requirement of the Nasdaq Capital Market.
Authorized Capital Stock
We are currently authorized to issue up to 25,000,000 shares of Common Stock, par value $0.007 per share, and 20,000,000 shares of Preferred Stock, par value $0.001 per share.
As of September 20, 2023, there were 9,350,297 shares of Common Stock, including 133,414 shares of unvested shares of restricted stock issued and outstanding that were held of record by 74 stockholders. As permitted by the Company’s Charter, the Company has designated 5,400,000 shares of Preferred Stock as “Series A Convertible Preferred Stock,” par value $0.001 per share (the “Series A Preferred Stock”), of which no shares are outstanding.
Common Stock
Voting Rights
Holders of our Common Stock are entitled to cast one vote for each share held of record on all matters presented to the stockholders. Holders of our Common Stock have no cumulative voting rights.
Dividend Rights
The Board is not obligated to declare a dividend, has never declared or paid cash dividends on its Common Stock, and does not anticipate paying dividends on our Common Stock for the foreseeable future.
Rights upon Liquidation
In the event of our liquidation, dissolution, or winding up, either voluntary or involuntary, subject to the rights and preferences that may apply to any shares of Preferred Stock outstanding at the time, the assets or surplus funds legally available for distribution to our stockholders would be distributable ratably among the Common Stockholders based on the number of shares of Common Stock held by each such holder, subject to prior satisfaction of all outstanding debt and liabilities.
No Preemptive or Similar Rights
Holders of our Common Stock are not entitled to preemptive rights to subscribe to additional shares if issued. Our Common Stock is not subject to any redemption or sinking-fund provisions. All outstanding shares of our Common Stock are fully paid and non-assessable.
Series A Preferred Stock
Voting Rights
Holders of the shares of Series A Preferred Stock have the right to one vote for each share of Common Stock into which such Series A Preferred Stock could then be converted. In addition, for so long as 30% of the shares of Series A Preferred Stock remain outstanding, the Series A Preferred Stock holders, voting together as a single class, may exercise the Series A Director Designation Right, pursuant to which they are entitled to elect one director of the Company as the Series A Representative. Any Series A Representative elected by the holders of Series A Preferred Stock may be removed from office only by the Series A Preferred Stock holders, and any vacancy of a Series A Representative may be filled only by the holders of the Series A Preferred Stock. If at any time fewer than 30% of the shares of Series A Preferred Stock remain outstanding, then the director position previously held by the Series A Representative will be elected by all of the holders of Preferred Stock and Common Stock acting together.
| 98 |
Dividend Rights
Holders of shares of the Series A Preferred Stock are entitled to receive dividends, in preference to any declaration or payment of a dividend to holders of the Common Stock, of 8% per share per annum when, as and if declared by the Board. Such dividends are not cumulative. See “Description of Securities—Dividend Policy” below.
Rights Upon Liquidation
In the event of any liquidation, dissolution or similar event, the holders of shares of Series A Preferred Stock are entitled to receive in preference to any distribution of any of the assets of the Company to the holders of the Common Stock, $7.70 per share (subsequent to the reverse-stock-split calculation). Unless otherwise decided by holders of a majority of the Preferred Stock outstanding, a liquidation includes a sale of substantially all of the assets of the Company and a merger, unless such merger is solely for the purpose of changing the Company’s state of incorporation or a majority of the voting power of the surviving entity will be owned by persons who were stockholders of the Company prior to the merger. Holders of shares of Preferred Stock will not participate with the holders of Common Stock in the distribution of the remainder of the Company’s assets.
Conversion Rights
Shares of Series A Preferred Stock are convertible, at the option of the holder thereof, into shares of Common Stock at any time. Shares of Series A Preferred Stock are automatically converted into shares of Common Stock following the closing of an underwritten initial public offering of our Common Stock in which at least $10,000,000 in shares of Common Stock are sold at a price of $3.00 per share or more or such other date as agreed to by a holders of the majority of the outstanding shares of Series A Preferred Stock. The holders of a more than a majority of our outstanding shares of Series A Preferred Stock executed a written consent such that all of the issued and outstanding shares of Series A Preferred Stock automatically converted into fully paid and nonassessable shares of Common Stock immediately prior to the closing of the initial public offering at the then-effective conversion rate of the Series A Preferred Stock. The conversion rate of Series A Preferred Stock into Common Stock is initially 1 for 7 (as adjusted for the 1-for-7 reverse stock split) but is subject to further adjustment in the event of a stock split, stock dividend or similar event.
Following the automatic conversion of the Series A Preferred Stock shares into Common Stock immediately prior to the closing of our initial public offering, the Company does not intend to issue any further shares of Series A Preferred Stock. Furthermore, the Series A Director Designation Right ceased to exist because no shares of Series A Preferred Stock are outstanding. The director who currently serves as the Series A Representative, Gary Rubin, will continue to serve as a director until his earlier resignation or removal or until his successor is duly elected and qualified. The number of Board seats for election by the holders of the Common Stock will be expanded by one so that the director position that the holders of the Series A Preferred Stock were previously entitled to elect will be subject to election the holders of the Common Stock following the conversion of the Series A Preferred Stock into Common Stock.
Warrants
Warrants Issued In Connection with Our Initial Public Offering
In connection with our initial public offering, we sold Tradeable Warrants to purchase up to 1,392,767 shares of our Common Stock at an exercise price equal to $7.35, of which 667,191 remained outstanding as of September 15, 2023 and Non-Tradeable Warrants to purchase up to 1,392,767 shares of our Common Stock at an exercise price equal to $7.66, of which 1,081,857 remained outstanding as of September 15, 2023. As of the date of this prospectus and upon consummation of the Acquisition, there are an aggregate of 4,305,812 shares of Common Stock issuable upon the exercise of outstanding Tradeable Warrants and Non-Tradeable Warrants, all of which have an exercise price of $3.0625 per share and expire five years from the date of issuance. The Tradeable Warrants are currently listed on Nasdaq and trade under the symbol “BIAFW.” This summary of certain terms and provisions of the warrants offered in our initial public offering is not complete and is subject to, and qualified in its entirety by the provisions of the form of warrants, which is filed as an exhibit to the registration statement of which this prospectus forms a part.
In connection with the sale of our convertible bridge notes, we issued Placement Agent’s Warrants to the Placement Agent, or their designees, to purchase an aggregate of 54,464 shares of our Common Stock. The Placement Agent’s Warrants have a five-year term commencing 180 days after the commencement of the sales in the initial public offering and have an exercise price of $7.35 per share.
In connection with our initial public offering, we agreed to issue to the representative of the underwriters or their designees warrants to purchase an aggregate of 26,652 shares of our Common Stock as additional consideration to the underwriters. The underwriters’ warrants have an exercise price of $7.044 per share, are exercisable during the period commencing 180 days after the commencement of sales in our initial public offering and expire five years from the effective date of our initial public offering and contain customary “cashless” exercise and registration rights provisions.
Other Outstanding Warrants
We also have outstanding warrants to purchase up to 2,900,904 shares of our Common Stock, having a weighted average exercise price of $5.31 expiring between October 14, 2026 and July 25, 2027, issued prior to consummation of our initial public offering.
Stock Options
As of the date of this prospectus, we had reserved the following shares of common stock for issuance:
| ● | 806,392 shares of our Common Stock reserved for issuance under stock option agreements with a weighted average exercise price of $4.33 per share; and | |
| ● | 658,294 shares of our common stock reserved for future issuance under the Plan. |
Exchange Listing.
Our Common Stock and the Tradeable Warrants trade on The Nasdaq Capital Market under the symbols “BIAF” and “BIAFW,” respectively.
| 99 |
Dividend Policy
We have never declared or paid cash dividends on our capital stock. We currently intend to retain all available funds and any future earnings for use in the operation and expansion of our business. We do not anticipate paying any cash dividends in the foreseeable future, and it is unlikely that investors will derive any current income from ownership of our stock.
Anti-Takeover Effects of Delaware Law and Provisions of Our Charter and A&R Bylaws
Certain provisions of the DGCL and of our Charter and our A&R Bylaws could have the effect of delaying, deferring or discouraging another party from acquiring control of us. These provisions, which are summarized below, are expected to discourage certain types of coercive takeover practices and inadequate takeover bids and, as a consequence, they might also inhibit temporary fluctuations in the market price of our Common Stock that often result from actual or rumored hostile takeover attempts. These provisions are also designed in part to encourage anyone seeking to acquire control of us to first negotiate with our Board. These provisions might also have the effect of preventing changes in our Board or management. It is possible that these provisions could make it more difficult to accomplish transactions that stockholders might otherwise deem to be in their best interests. However, we believe that the advantages gained by protecting our ability to negotiate with any unsolicited and potentially unfriendly acquirer outweigh the disadvantages of discouraging such proposals, including those priced above the then-current market value of our Common Stock, because, among other reasons, the negotiation of such proposals could improve their terms.
Delaware Anti-Takeover Statute
We are subject to the provisions of Section 203 of the DGCL. In general, Section 203 prohibits a publicly held Delaware corporation from engaging in a “business combination” with an “interested stockholder” for a three-year period following the time that this stockholder becomes an interested stockholder, unless the business combination is approved in a prescribed manner. Under Section 203, a business combination between a corporation and an interested stockholder is prohibited unless it satisfies one of the following conditions:
| ● | before the stockholder became interested, the corporation’s board of directors approved either the business combination or the transaction that resulted in the stockholder becoming an interested stockholder; | |
| ● | upon consummation of the transaction that resulted in the stockholder becoming an interested stockholder, the interested stockholder owned at least 85% of the voting stock of the corporation outstanding at the time the transaction commenced, excluding for purposes of determining the voting stock outstanding, shares owned by persons who are directors and also officers, and employee stock plans, in some instances, but not the outstanding voting stock owned by the interested stockholder; or | |
| ● | at or after the time the stockholder became interested, the business combination was approved by the corporation’s board of directors and authorized at an annual or special meeting of the stockholders by the affirmative vote of at least two-thirds of the outstanding voting stock that is not owned by the interested stockholder. |
In general, Section 203 defines a “business combination” to include mergers, asset sales and other transactions resulting in financial benefit to a stockholder, and an “interested stockholder” as a person who, together with affiliates and associates, owns, or within three years did own, 15% or more of the corporation’s outstanding voting stock. These provisions may have the effect of delaying, deferring or preventing changes in control of our Company.
Provisions of Our Charter and A&R Bylaws
Our Charter and A&R Bylaws include a number of provisions that may have the effect of delaying, deferring, or discouraging another party from acquiring control of us and encouraging persons considering unsolicited tender offers or other unilateral takeover proposals to negotiate with our Board rather than pursue non-negotiated takeover attempts. These provisions will include the items described below.
Director Vacancies
Our A&R Bylaws authorize the Board to fill vacant directorships and provide that the number of directors constituting our Board may be set by resolution of the incumbent directors.
Special Meetings of Stockholders
Our A&R Bylaws provide that special meetings of our stockholders may only be called pursuant to a resolution approved by the Board. The only business that may be conducted at a special meeting of our stockholders is the matter or matters set forth in the notice of such special meeting.
Prohibition of Stockholder Action by Written Consent
Our Charter and A&R Bylaws prohibit stockholder action by written consent, thereby requiring all stockholder actions to be taken at a meeting of our stockholders.
Advance Notice Requirements
Our A&R Bylaws establish advance notice requirements for nominations for election to the board of directors or for proposing matters that can be acted upon at stockholder meetings. To be timely, a stockholder’s notice will need to be received by the Company secretary at our principal executive offices (x) not later than the close of business on the 90th day nor earlier than the close of business on the 120th day prior to the anniversary date of the immediately preceding annual meeting of stockholders (if such meeting is to be held on a day which is not more than 30 days in advance of the anniversary of the previous year’s annual meeting or not later than 60 days after the anniversary of the previous year’s annual meeting), or (y) with respect to any other annual meeting of stockholders, including in the event that no annual meeting was held in the previous year, not earlier than the close of business on the 120th day prior to the annual meeting and not later than the close of business on the later of: (1) the 90th day prior to the annual meeting and (2) the close of business on the tenth day following the first date that the date of such meeting was disclosed in a press release reported by the Dow Jones News Services, The Associated Press, or a comparable national news service or in a document filed by the Company with the SEC pursuant to the Exchange Act. Our A&R Bylaws also specify certain requirements as to the form and content of a stockholders’ meeting. These provisions may preclude our stockholders from bringing matters before our annual meeting of stockholders or from making nominations for directors at our annual meeting of stockholders.
| 100 |
Amendment to Charter and Bylaws
As required by the DGCL, any amendment of our Charter must first be approved by a majority of our Board, and if required by law or our Charter, must thereafter be approved by a majority of the outstanding shares entitled to vote on the amendment, and a majority of the outstanding shares of each class entitled to vote thereon as a class. Our A&R Bylaws provide for amendment of the A&R Bylaws by a majority of our Board or by a majority of the outstanding shares entitled to vote on the amendment.
Exclusive Forum
Both our Charter and our A&R Bylaws contain exclusive forum provisions that provide that unless we consent in writing to the selection of an alternative forum, the Court of Chancery of the State of Delaware shall be the sole and exclusive forum for any stockholder to bring (i) any derivative action or proceeding brought on behalf of the Company, (ii) any action asserting a claim of breach of fiduciary duty owed by any current or former director, officer, employee or agent of the Company to the Company or the Company’s stockholders, (iii) any action asserting a claim arising pursuant to the DGCL, our Charter or A&R Bylaws (as either may be amended or restated) or as to which the DGCL confers jurisdiction on the Court of Chancery of the State of Delaware, or (iv) any action asserting a claim governed by the internal affairs doctrine of the State of Delaware. These provisions expressly do not apply to claims arising under the Exchange Act, or for any other federal securities laws which provide for exclusive federal jurisdiction. However, these exclusive forum provisions provide that unless we consent in writing to the selection of an alternative forum, the federal district courts of the United States of America will be the exclusive forum for the resolution of any complaint asserting a cause of action arising under the Securities Act. Therefore, this provision could apply to a suit that falls within one or more of the categories enumerated in the exclusive forum provision and that asserts claims under the Securities Act, inasmuch as Section 22 of the Securities Act creates concurrent jurisdiction for federal and state courts over all suits brought to enforce any duty or liability created by the Securities Act or the rules and regulations thereunder. There is uncertainty as to whether a court would enforce such an exclusive forum provision with respect to claims under the Securities Act. Stockholders cannot waive compliance with the federal securities laws and the rules and regulations thereunder. Any person or entity purchasing or otherwise acquiring or holding any interest in shares of our capital stock shall be deemed to have notice of and consented to the exclusive forum provisions in our Charter and A&R Bylaws. These exclusive forum provisions may limit a stockholder’s ability to bring a claim in a judicial forum of its choosing for disputes with us or our directors, officers, employees, or agents, which may discourage lawsuits against us and our directors, officers, employees, and agents.
Limitations on Liability and Indemnification of Officers and Directors
For a discussion of liability and indemnification, see the section entitled “Management—Limitations on Liability and Indemnification of Officers and Directors.”
| 101 |
DESCRIPTION OF SECURITIES WE ARE OFFERING
We are offering 3,592,814 Units comprised of up to 3,592,814 shares of our Common Stock and Warrants to purchase up to 3,592,814 shares of our Common Stock. No Warrant for fractional shares of common stock will be issued, rather warrants will be issued only for whole shares of Common Stock. We are also registering the shares of Common Stock issuable from time to time upon exercise of the Warrants offered hereby.
Common Stock
The material terms and provisions of our Common Stock are described under the caption “Description of Securities” in this prospectus.
Warrants to Be Issued in the Offering
Overview. The following summary of certain terms and provisions of the Warrants included in the Units offered hereby is not complete and is subject to, and qualified in its entirety by, the provisions of the Warrant Agent Agreement between us and VStock Transfer, LLC, as Warrant Agent, and the form of Warrant, all of which are filed as exhibits to this prospectus. Prospective investors should carefully review the terms and provisions set forth in the Warrant Agent Agreement, including the annexes thereto, and form of Warrant.
Exercisability. The Warrants are exercisable at any time after their original issuance and at any time up to the date that is five years after their original issuance. The Warrants will be exercisable, at the option of each holder, in whole or in part by delivering to us a duly executed exercise notice and, at any time a registration statement registering the issuance of the shares of Common Stock underlying the Warrants under the Securities Act is effective and available for the issuance of such shares, or an exemption from registration under the Securities Act is available for the issuance of such shares, by payment in full in immediately available funds for the number of shares of Common Stock purchased upon such exercise. If a registration statement registering the issuance of the shares of Common Stock underlying the Warrants under the Securities Act is not effective or available and an exemption from registration under the Securities Act is not available for the issuance of such shares, the holder of a Warrant may, in its sole discretion, elect to exercise the Warrant through a cashless exercise, in which case the holder would receive upon such exercise the net number of shares of Common Stock determined according to the formula set forth in the Warrant. In addition, if the Warrants are not exercised prior to their termination date, they will be automatically exercised via a cashless exercise on their termination date.
Exercise Limitation. A holder of a Warrant will not have the right to exercise any portion of the Warrant if the holder (together with its affiliates and any other person or entity acting as a group) would beneficially own more than 4.99% of the number of shares of our Common Stock outstanding immediately after giving effect to the exercise, as such percentage ownership is determined in accordance with the terms of the Warrants. However, upon notice from the holder to us, the holder may waive such limitation up to a percentage, not in excess of 9.99%, provided that any increase in such percentage shall not be effective until 61 days following delivery of such notice from the holder to us.
Exercise Price. The exercise price per whole share of Common Stock purchasable upon exercise of the Warrant is $[●] per share and is subject to appropriate adjustment in the event of certain stock dividends and distributions, stock splits, stock combinations, reclassifications or similar events affecting our Common Stock.
Fractional Shares. No fractional shares of Common Stock will be issued upon exercise of the Warrants. If, upon exercise of a Warrant, a holder would be entitled to receive a fractional interest in a share, we will, upon exercise, round up to the next whole share.
Transferability. Subject to applicable laws, the Warrants may be offered for sale, sold, transferred or assigned without our consent.
Warrant Agent; Global Certificates. The Warrants will be issued in registered form under a Warrant Agent Agreement between the Warrant Agent and us. The Warrants shall initially be represented only by one or more global warrants deposited with the Warrant Agent, as custodian on behalf of The Depository Trust Company (“DTC”) and registered in the name of Cede & Co., a nominee of DTC, or as otherwise directed by DTC, but the holder may request that some or all of such holder’s Global Warrants be exchanged for a separate “Definitive Certificate.” Our transfer agent, VStock Transfer, LLC, will serve as the Warrant Agent.
Fundamental Transactions. In the event of a fundamental transaction, as described in the Warrants and generally including any reorganization, recapitalization or reclassification of our Common Stock, the sale, transfer or other disposition of all or substantially all of our properties or assets, our consolidation or merger with or into another person, or the acquisition of more than 50% of our outstanding Common Stock, the holders of the Warrants will be entitled to receive upon exercise of the Warrants the kind and amount of securities, cash or other property that the holders would have received had they exercised the Warrants immediately prior to such fundamental transaction. Notwithstanding the foregoing, in the event of a fundamental transaction, the holders of the Common Warrants have the right to require us or a successor entity to redeem the warrants for cash in the amount of the Black-Scholes Value (as defined in each warrant) of the unexercised portion of the Common Warrants concurrently with or within 30 days following the consummation of a fundamental transaction. However, in the event of a fundamental transaction which is not in our control, including a fundamental transaction not approved by our board of directors, the holders of the Common Warrants will only be entitled to receive from us or our successor entity, as of the date of consummation of such fundamental transaction the same type or form of consideration (and in the same proportion), at the Black Scholes Value of the unexercised portion of the Common Warrant that is being offered and paid to the holders of our common stock in connection with the fundamental transaction, whether that consideration is in the form of cash, stock or any combination of cash and stock, or whether the holders of our common stock are given the choice to receive alternative forms of consideration in connection with the fundamental transaction
Rights as a Stockholder. Except as otherwise provided in the Warrants or by virtue of such holder’s ownership of shares of our Common Stock, the holder of a Warrant does not have the rights or privileges of a holder of our Common Stock, including any voting rights, until the holder exercises the Warrant.
Cashless Exercise. If at the time of exercise there is no effective registration statement registering the issuance of the Warrant Shares, then the holder of a Warrant may, in its sole discretion, exercise in whole or in part, and in lieu of making the cash payment otherwise contemplated to be made to the Company upon such exercise in payment of the aggregate exercise price, elect instead to exercise the Warrant on a cashless basis. Notwithstanding anything herein to the contrary, the Company shall not be required to make any cash payments or net cash settlement to the Warrant holder in lieu of delivery of the Warrant Shares. Upon a “cashless exercise,” the Warrant holder shall be entitled to receive the number of Warrant Shares equal to the quotient obtained by dividing (A-B) (X) by (A), where:
(A) = the last VWAP immediately preceding the date of exercise giving rise to the applicable “cashless exercise,” as set forth in the applicable Election to Purchase (as defined in the Warrant Agent Agreement) (to clarify, the “last VWAP” will be the last VWAP as calculated over an entire trading day such that, in the event that the Warrant is exercised at a time that the trading market is open, the prior trading day’s VWAP shall be used in this calculation);
| 102 |
(B) = the Exercise Price then in effect for the applicable Warrant Shares at the time of the exercise of the Warrant, as adjusted as set forth herein; and
(X) = the number of Warrant Shares that would be issuable upon exercise of the Warrant in accordance with the terms of the Warrant if such exercise were by means of a cash exercise rather than a cashless exercise.
If the Warrants are not exercised prior to their termination date, they will be automatically exercised via a cashless exercise on their termination date.
Governing Law; and Exclusive Forum. The Warrants and the Warrant Agent Agreement are governed by New York law. The warrant certificates governing the Warrants provide that all legal proceedings concerning the interpretations, enforcement and defense of the transactions contemplated by the warrant certificate (whether brought against a party to the warrant certificate or their respective affiliates, directors, officers, shareholders, partners, members, employees or agents) shall be commenced exclusively in the state and federal courts sitting in the City of New York, Borough of Manhattan. The warrant certificates further provide that we and the Warrant holders irrevocably submit to the exclusive jurisdiction of the state and federal courts sitting in the City of New York, Borough of Manhattan, for the adjudication of any dispute under the warrant certificate or in connection with it or with any transaction contemplated by it or discussed in it. Furthermore, we and the Warrant holders irrevocably waive, and agree not to assert in any suit, action or proceeding, any claim that we or they are not personally subject to the jurisdiction of any such court, that such suit, action or proceeding is improper or is an inconvenient venue for such proceeding. With respect to any complaint asserting a cause of action arising under the Securities Act or the rules and regulations promulgated thereunder, we note, however, that there is uncertainty as to whether a court would enforce this provision and that investors cannot waive compliance with the federal securities laws and the rules and regulations thereunder. Section 22 of the Securities Act creates concurrent jurisdiction for state and federal courts over all suits brought to enforce any duty or liability created by the Securities Act or the rules and regulations thereunder. Section 27 of the Exchange Act creates exclusive federal jurisdiction over all suits brought to enforce any duty or liability created by the Exchange Act or the rules and regulations thereunder. As a result, the exclusive forum provision in the Warrant certificates expressly does not apply to suits brought to enforce any duty or liability created by the Exchange Act. We irrevocably waive any right we may have to, and agree not to request, a jury trial for the adjudication of any dispute under, in connection with, or arising out of the Warrant or any transaction contemplated by the Warrant.
Representative’s Warrant
As additional compensation to the underwriters, upon consummation of this Offering, we will issue to the Representative or its designees a non-redeemable Representative’s Warrant to purchase an aggregate number of shares of our Common Stock equal to two percent (2.0%) of the number of shares of Common Stock underlying the Units issued in this Offering, at an exercise price per share equal to $[●], which may be via a “cashless exercise.”
The Representative’s Warrant will be exercisable, in whole or in part, commencing on the six-month anniversary of the commencement of the sales of the public securities in this Offering and will expire on the fifth anniversary of the effective date of the registration statement related to the Offering. In addition, we have granted the underwriters the ability to exercise them in a “cashless” manner, a one-time demand registration right at our expense, an additional demand registration at the holder’s expense, and unlimited “piggyback” registration rights with respect to the underlying shares. See “Underwriting—Representative’s Warrant.”
| 103 |
MATERIAL U.S. FEDERAL INCOME TAX CONSIDERATIONS TO NON-U.S. HOLDERS
The following discussion is a summary of the material U.S. federal income tax considerations applicable to Non-U.S. Holders (as defined below) with respect to their acquisition, ownership and disposition of shares of our Common Stock underlying the Units issued pursuant to this Offering. This summary does not provide a complete analysis of all potential U.S. federal income tax considerations relating thereto. The information provided below is based upon provisions of the U.S. Internal Revenue Code of 1986, as amended (the “Code”), Treasury regulations promulgated thereunder, administrative rulings, and judicial decisions currently in effect. These authorities may change at any time, possibly retroactively, or the Internal Revenue Service (the “IRS”) might interpret the existing authorities differently. In either case, the tax considerations of owning or disposing of our Common Stock could differ from those described below. As a result, we cannot assure you that the tax consequences described in this discussion will not be challenged by the IRS or will be sustained by a court if challenged by the IRS.
This summary does not address the tax considerations arising under the laws of any non-U.S., state or local jurisdiction, or under U.S. federal gift and estate tax laws, except to the limited extent provided below. In addition, this discussion does not address tax considerations applicable to an investor’s particular circumstances or to investors that may be subject to special tax rules, including, without limitation:
| ● | banks, insurance companies or other financial institutions; | |
| ● | partnerships or entities or arrangements treated as partnerships or other pass-through entities for U.S. federal tax purposes (or investors in such entities); | |
| ● | corporations that accumulate earnings to avoid U.S. federal income tax; | |
| ● | persons subject to the alternative minimum tax or Medicare contribution tax on net investment income; | |
| ● | tax-exempt organizations or tax-qualified retirement plans; | |
| ● | controlled foreign corporations or passive foreign investment companies; | |
| ● | dealers in securities or currencies; | |
| ● | traders in securities that elect to use a mark-to-market method of accounting for their securities holdings; | |
| ● | persons that own, or are deemed to own, more than 5% of our capital stock (except to the extent specifically set forth below); | |
| ● | certain former citizens or former long-term residents of the United States; | |
| ● | persons who hold our Common Stock as a position in a hedging transaction, “straddle,” “conversion transaction” or other risk reduction transaction; | |
| ● | persons who do not hold our Common Stock as a capital asset within the meaning of Section 1221 of the Code (generally, for investment purposes); or | |
| ● | persons deemed to sell our Common Stock under the constructive sale provisions of the Code. |
In addition, if a partnership or entity classified as a partnership for U.S. federal income tax purposes is a beneficial owner of our Common Stock, the tax treatment of a partner in the partnership or an owner of the entity will depend upon the status of the partner or other owner and the activities of the partnership or other entity. Accordingly, this summary does not address tax considerations applicable to partnerships that hold our Common Stock, and partners in such partnerships should consult their tax advisors.
INVESTORS CONSIDERING THE PURCHASE OF OUR COMMON STOCK SHOULD CONSULT THEIR OWN TAX ADVISORS REGARDING THE APPLICATION OF THE U.S. FEDERAL INCOME AND ESTATE TAX LAWS TO THEIR PARTICULAR SITUATIONS AND THE CONSEQUENCES OF FOREIGN, STATE OR LOCAL LAWS, AND TAX TREATIES.
Non-U.S. Holder Defined
For purposes of this summary, a Non-U.S. Holder is any beneficial owner of our Common Stock, other than a partnership, that is not:
| ● | an individual who is a citizen or resident of the United States; | |
| ● | a corporation, or other entity taxable as a corporation for U.S. federal income tax purposes, created or organized under the laws of the United States, any state therein or the District of Columbia; | |
| ● | a trust if it (i) is subject to the primary supervision of a U.S. court and one of more U.S. persons have authority to control all substantial decisions of the trust or (ii) has a valid election in effect under applicable U.S. Treasury regulations to be treated as a U.S. person; or | |
| ● | an estate whose income is subject to U.S. income tax regardless of source. |
If you are a non-U.S. citizen that is an individual, you may, in many cases, be treated as a resident alien, as opposed to a nonresident alien, by virtue of being present in the United States for at least 31 days in the calendar year and for an aggregate of at least 183 days during a three-year period ending in the current calendar year. For these purposes, all the days present in the current year, one-third of the days present in the immediately preceding year, and one-sixth of the days present in the second preceding year are counted. Resident aliens are subject to U.S. federal income tax as if they were U.S. citizens. Such an individual is urged to consult his or her own tax advisor regarding the U.S. federal income tax consequences of the ownership or disposition of our Common Stock.
| 104 |
Dividends
As discussed under “Dividend Policy” above, we do not currently expect to declare or pay dividends to our Common Stockholders in the foreseeable future. In the event that we do make distributions of cash or other property on our Common Stock, those distributions will constitute dividends for U.S. federal income tax purposes to the extent paid from our current or accumulated earnings and profits, as determined under U.S. federal income tax principles. Amounts not treated as dividends for U.S. federal income tax purposes will constitute a return of capital, which will first reduce a Non-U.S. Holder’s adjusted tax basis in shares of our Common Stock, but not below zero. Any remaining excess will be treated as gain realized on the sale or other disposition of our Common Stock and will be treated as described below under “Gain on Sale or Other Taxable Disposition of Our Common Stock.”
Subject to the discussion below on effectively connected income, dividends paid to a Non-U.S. Holder of our Common Stock that is not effectively connected with the Non-U.S. Holder’s conduct of a trade or business in the United States will generally be subject to U.S. federal withholding tax at a rate of 30% of the gross amount of the dividends (or such lower rate specified by an applicable income tax treaty, provided the Non-U.S. Holder furnishes a properly executed IRS Form W-8BEN or W-8BEN-E (or other applicable or successor form) certifying the Non-U.S. Holder’s qualification for the lower treaty rate). A Non-U.S. Holder that does not timely furnish the required documentation, but that qualifies for a reduced treaty rate, may obtain a refund of any excess amounts withheld by timely filing an appropriate claim for refund with the IRS. Non-U.S. Holders should consult their tax advisors regarding their entitlement to benefits under any applicable income tax treaty. If the Non-U.S. Holder holds the stock through a financial institution or other agent acting on the holder’s behalf, the holder will be required to provide appropriate documentation to the agent. The holder’s agent will then be required to provide certification to us or our paying agent, either directly or through other intermediaries. If you are eligible for a reduced rate of U.S. federal withholding tax under an income tax treaty, you may obtain a refund or credit of any excess amounts withheld by filing an appropriate claim for a refund with the IRS in a timely manner.
If dividends paid to a Non-U.S. Holder are effectively connected with the Non-U.S. Holder’s conduct of a trade or business in the United States (and, if required by an applicable income tax treaty, are attributable to a permanent establishment or fixed base maintained by the Non-U.S. Holder in the United States), the Non-U.S. Holder will be exempt from the U.S. federal withholding tax described above. To claim the exemption, the Non-U.S. Holder must furnish to the applicable withholding agent a valid IRS Form W-8ECI, certifying that the dividends are effectively connected with the Non-U.S. Holder’s conduct of a trade or business within the United States.
Any such effectively connected dividends will be subject to U.S. federal income tax on a net income basis at the regular rates. A Non-U.S. Holder that is a corporation also may be subject to a branch profits tax at a rate of 30% (or such lower rate specified by an applicable income tax treaty) on such effectively connected dividends, as adjusted for certain items. Non-U.S. Holders should consult their tax advisors regarding any applicable tax treaties that may provide for different rules.
Gain on Sale or Other Taxable Disposition of Our Common Stock
Subject to the discussion below under “Information Reporting and Backup Withholding” and “Foreign Account Tax Compliance Act,” a Non-U.S. Holder will generally not be subject to U.S. federal income tax on any gain realized upon the sale, exchange, or other taxable disposition of our Common Stock unless:
| ● | the gain (i) is effectively connected with the conduct by the Non-U.S. Holder of a U.S. trade or business, and (ii) if required by an applicable income tax treaty between the United States and the Non-U.S. holder’s country of residence, is attributable to a permanent establishment maintained by the Non-U.S. Holder in the United States (in which the special rules described below apply); | |
| ● | the Non-U.S. Holder is an individual who is present in the United States for 183 days or more in the taxable year of the sale, exchange or other disposition of our Common Stock, and certain other requirements are met (in which case the gain would be subject to a flat 30% tax, or such reduced rate as may be specified by an applicable income tax treaty, which may be offset by certain U.S. source capital losses, even though the individual is not considered a resident of the United States); or | |
| ● | the rules of the Foreign Investment in Real Property Tax Act (“FIRPTA”) treat the stock as a “U.S. real property interest” as defined in Section 897 of the Code. |
The FIRPTA rules may apply to a sale, exchange or other disposition of our Common Stock if we are, or were within the shorter of the five-year period preceding the disposition and the Non-U.S. Holder’s holding period, a “U.S. real property holding corporation” (a “USRPHC”), as defined in Section 897 of the Code. In general, we would be a USRPHC if interests in U.S. real estate comprised at least half of the value of our business assets. We do not believe that we are a USRPHC and we do not anticipate becoming one in the future. Even if we become a USRPHC, as long as our Common Stock is regularly traded on an established securities market, such Common Stock will be treated as U.S. real property interests only if beneficially owned by a Non-U.S. Holder that actually or constructively owned more than 5% of our outstanding Common Stock at sometime within the five-year period preceding the disposition.
If any gain from the sale, exchange or other disposition of our Common Stock (1) is effectively connected with a U.S. trade or business conducted by a Non-U.S. Holder, and (2) if required by an applicable income tax treaty between the United States and the Non-U.S. Holder’s country of residence, is attributable to a permanent establishment maintained by such Non-U.S. Holder in the United States, then the gain generally will be subject to U.S. federal income tax at the same graduated rates applicable to U.S. persons, net of certain deductions and credits. If the Non-U.S. Holder is a corporation, under certain circumstances, that portion of its earnings and profits that is effectively connected with its U.S. trade or business, subject to certain adjustments, generally would be subject also to a “branch profits tax.” The branch profits tax rate is 30% unless reduced by applicable income tax treaty.
Non-U.S. Holders should consult their tax advisors regarding potentially applicable income tax treaties that may provide for different rules.
U.S. Federal Estate Tax
The estates of nonresident alien individuals generally are subject to U.S. federal estate tax on property with a U.S. situs. Because we are a U.S. corporation, our Common Stock will be U.S. situs property and therefore will be included in the taxable estate of a nonresident alien decedent, unless an applicable estate tax treaty between the United States and the decedent’s country of residence provides otherwise.
| 105 |
Informational Reporting and Backup Withholding
The Code and the Treasury regulations require those who make specified payments to report the payments to the IRS. Among the specified payments are dividends and proceeds paid by brokers to their customers. The required information returns enable the IRS to determine whether the recipient properly included the payments in income. This reporting regime is reinforced by “backup withholding” rules. These rules require the payors to withhold tax from payments subject to information reporting if the recipient fails to cooperate with the reporting regime by failing to provide his taxpayer identification number to the payor, furnishing an incorrect identification number, or failing to report interest or dividends on his returns. The backup withholding tax rate is currently 24%. The backup withholding rules do not apply to payments to corporations, whether domestic or foreign, provided they establish such exemption.
Payments to Non-U.S. Holders of dividends on our Common Stock generally will not be subject to backup withholding, and payments of proceeds made to Non-U.S. Holders by a broker upon a sale of Common Stock will not be subject to information reporting or backup withholding, in each case so long as the Non-U.S. Holder certifies its status as a Non-U.S. Holder (and we or our paying agent do not have actual knowledge or reason to know the holder is a U.S. person or that the conditions of any other exemption are not, in fact, satisfied) or otherwise establishes an exemption. The certification procedures to claim treaty benefits described under “Distributions” will generally satisfy the certification requirements necessary to avoid the backup withholding tax. We must report annually to the IRS any dividends paid to each Non-U.S. Holder and the tax withheld, if any, with respect to these dividends. Copies of these reports may be made available to tax authorities in the country where the Non-U.S. Holder resides. However, under the Treasury regulations, information returns are required to be filed with the IRS in connection with any dividends on our Common Stock paid to the Non-U.S. Holder, regardless of whether any tax was actually withheld. In addition, proceeds of the sale or other taxable disposition of our Common Stock within the United States or conducted through certain U.S.-related brokers generally will not be subject to backup withholding or information reporting, if the beneficial owner certifies, under penalties of perjury, among other things, its status as a Non-U.S. Holder (and the broker does not have actual knowledge or reason to know the holder is a U.S. person) or otherwise establishes an exemption. The payment of proceeds from the disposition of shares of our Common Stock by a Non-U.S. Holder made to or through a non-U.S. office of a broker generally will not be subject to backup withholding and information reporting, except as noted below. Information reporting, but not backup withholding, will apply to a payment of proceeds, even if that payment is made outside of the United States, if you sell our Common Stock through a non-U.S. office of a broker that is:
| ● | a U.S. person (including a foreign branch or office of such person); | |
| ● | a “controlled foreign corporation” for U.S. federal income tax purposes; | |
| ● | a foreign person 50% or more of whose gross income from certain periods is effectively connected with a U.S. trade or business; or | |
| ● | a foreign partnership if at any time during its tax year (a) one or more of its partners are U.S. persons who, in the aggregate, hold more than 50% of the income or capital interests of the partnership or (b) the foreign partnership is engaged in a U.S. trade or business, unless the broker has documentary evidence that the beneficial owner is a Non-U.S. Holder and certain other conditions are satisfied, or the beneficial owner otherwise establishes an exemption (and the broker has no actual knowledge or reason to know to the contrary). |
Backup withholding is not an additional tax. Any amounts withheld under the backup withholding rules may be allowed as a refund or a credit against a Non-U.S. Holder’s U.S. federal income tax liability, provided the required information is timely furnished to the IRS.
Foreign Account Tax Compliance Act
A U.S. federal withholding tax of 30% may apply to dividends and the gross proceeds of a disposition of our Common Stock paid to a foreign financial institution (as specifically defined by the applicable rules) unless such institution enters into an agreement with the U.S. government to withhold on certain payments and to collect and provide to the U.S. tax authorities substantial information regarding U.S. account holders of such institution (which includes certain equity holders of such institution, as well as certain account holders that are foreign entities with U.S. owners). This U.S. federal withholding tax of 30% will also apply to dividends and the gross proceeds of a disposition of our Common Stock paid to a non-financial foreign entity unless such entity provides the withholding agent with either a certification that it does not have any substantial direct or indirect U.S. owners or provides information regarding direct and indirect U.S. owners of the entity. The 30% federal withholding tax described in this paragraph cannot be reduced under an income tax treaty with the United States or by providing an IRS Form W-8BEN or similar documentation. The withholding tax described above will not apply if the foreign financial institution or non-financial foreign entity otherwise qualifies for an exemption from the rules and certifies as such on a Form W-8BEN-E (or any successor of such form). Under certain circumstances, a Non-U.S. Holder might be eligible for refunds or credits of such taxes. Holders should consult with their own tax advisors regarding the possible implications of the withholding described herein.
The withholding provisions described above generally apply to proceeds from a sale or other disposition of Common Stock if such sale or other disposition occurs on or after January 1, 2019 and to payments of dividends on our Common Stock.
THE PRECEDING DISCUSSION OF U.S. FEDERAL TAX CONSIDERATIONS IS FOR GENERAL INFORMATION ONLY. IT IS NOT TAX ADVICE. EACH PROSPECTIVE INVESTOR SHOULD CONSULT ITS OWN TAX ADVISOR REGARDING THE PARTICULAR U.S. FEDERAL, STATE, LOCAL, AND FOREIGN TAX CONSEQUENCES OF PURCHASING, HOLDING AND DISPOSING OF OUR COMMON STOCK, INCLUDING THE CONSEQUENCES OF ANY PROPOSED CHANGE IN APPLICABLE LAWS.
| 106 |
UNDERWRITING
Under the terms and subject to the conditions in an underwriting agreement dated [●], 2023, the underwriters named below, for whom WallachBeth Capital, LLC is acting as the lead managing underwriter and sole book runner and the representative of the several underwriters (the “Representative”), have severally agreed to purchase, and we have agreed to sell to them, severally, the number of Units indicated below:
| Underwriters | Number
of Units |
|||
| WallachBeth Capital, LLC | [ ] | |||
| Craft Capital Management LLC | [ ] | |||
| Totals: | ||||
The underwriters are collectively referred to as the “underwriters,” and the Representative of the underwriters is WallachBeth Capital, LLC. The underwriters are offering the Units subject to their acceptance of the Units from us and subject to prior sale. The underwriting agreement provides that the obligations of the several underwriters to pay for and accept delivery of the Units offered by this prospectus are subject to the approval of certain legal matters by their counsel and to certain other conditions. The underwriters are obligated to take and pay for all of the Units offered by this prospectus if any such Units are taken. However, the underwriters are not required to take or pay for any of the Common Stock or Warrants covered by the underwriters’ Over-Allotment Option described below.
The underwriters initially propose to offer part of the Units directly to the public at the Offering Price listed on the cover page of this prospectus and part to certain dealers. After the initial offering of the Units, the offering price and other selling terms may from time to time be varied by the Representative.
Over-Allotment Option
We have granted to the underwriters an Over-Allotment Option, exercisable for 45 days from the date of this prospectus, to purchase up to an additional 462,962 shares of Common Stock, each share at the Offering Price per Unit $[●], Warrants at the price of $[●] per Warrant, or 15% of the total number of Units sold in the Offering in shares of Common Stock or in Warrants or any combination of shares of Common Stock or Warrants, less the underwriting discounts and commissions. The underwriters may exercise this option solely for the purpose of covering over-allotments, if any, made in connection with the Offering of the Units offered by this prospectus. To the extent the option is exercised, each underwriter will become obligated, subject to certain conditions, to purchase about the same percentage of the additional shares of Common Stock or Warrants as the number listed next to the underwriter’s name in the preceding table bears to the total number of Units listed next to the names of all underwriters in the preceding table.
Discounts and Commissions and Expenses
We have agreed to pay the underwriters a cash fee equal to nine percent (9.0%) of the aggregate gross proceeds from the sale of the Units.
The Representative has advised us that the underwriters propose to offer the Units directly to the public at the Offering Price set forth on the cover of this prospectus. In addition, the Representative may offer some of the Units to other securities dealers at such price less a concession of up to $[●] per Unit. After the Offering to the public, the offering price and other selling terms may be changed by the Representative without changing the Company’s proceeds from the underwriters’ purchase of the Units.
The following table shows the Offering Price, underwriting discounts and proceeds, before expenses, to us. The information assumes either no exercise or full exercise by the underwriters of their Over-Allotment Option. The underwriting discounts are equal to the Offering Price per Unit less the amount per Unit the underwriters pay us for the Units.
| Per Unit | Total
without Over-Allotment Option |
Total
with Over-Allotment Option |
||||||||||
| Public Offering Price | $ | $ | $ | |||||||||
| Underwriting discounts (9.0%)(1) | $ | $ | $ | |||||||||
| Proceeds, before expenses, to us | $ | $ | $ | |||||||||
We estimate that the total expenses of this Offering, including registration, filing, and listing fees, printing fees and legal and accounting expenses, will be approximately $[●]. This figure includes expense reimbursements we have agreed to pay the Representative for reimbursement of its expenses related to the Offering up to a maximum aggregate expense allowance of $145,000. In accordance with FINRA Rule 5110, the reimbursement fee described in the preceding sentence is deemed underwriting compensation for this Offering.
| 107 |
Representative’s Warrant
As additional compensation to the underwriters, upon consummation of this Offering, we will issue to the Representative or its designees a non-redeemable Representative’s Warrant to purchase an aggregate number of shares of our Common Stock equal to two percent (2.0%) of the number of shares of Common Stock underlying the Units issued in this Offering, at an exercise price per share equal to $[●], which is equal to 115% of the Offering Price, which may be via a “cashless exercise.” The Representative’s Warrant and the underlying shares of Common Stock shall not be sold during the Offering, or sold, transferred, assigned, pledged, or hypothecated, or be the subject of any hedging, short sale, derivative, put, or call transaction that would result in the effective economic disposition of the securities by any person for a period of six months immediately following the commencement of the sale of the public securities in accordance with FINRA Rule 5110(e)(1). The Representative’s Warrant will be exercisable, in whole or in part, commencing on the six month anniversary of the commencement of the sales of the public securities and will expire on the fifth anniversary of the effective date of the registration statement related to the Offering. In addition, we have granted the underwriters the ability to exercise them in a “cashless” manner, a one-time demand registration right at our expense, an additional demand registration at the holder’s expense, and unlimited “piggyback” registration rights with respect to the underlying shares. The demand registration rights will not be greater than five years from the effective date of the registration statement related to the Offering in compliance with FINRA Rule 5110(G)(8)(C). The piggyback registration rights will not be greater than seven (7) years from the effective date of the registration statement related to the Offering in compliance with FINRA Rule 5110(G)(8)(D).
Pricing of the Offering
Prior to this Offering, there has been no public market for our Common Stock. In determining the Offering Price, we and the Representative have considered a number of factors including:
| ● | the information set forth in this prospectus and otherwise available to the underwriters; | |
| ● | our prospects and the history and prospects for the industry in which we compete; | |
| ● | an assessment of our management; | |
| ● | our prospects for future earnings; | |
| ● | the general condition of the securities markets at the time of this Offering; | |
| ● | the recent market prices of, and demand for, publicly traded securities of generally comparable companies; and | |
| ● | other factors deemed relevant by the underwriters and us. |
Neither we nor the underwriters can assure investors that an active trading market will develop for shares of our Common Stock, or that the shares will trade in the public market at or above the Offering Price.
Lock-Up Agreements
The Company, on behalf of itself and any successor entity, has agreed that, without the prior written consent of the Representative, it will not, for a period of 180 days after the date of the underwriting agreement (the “Lock-Up Period”), without the Representative’s consent: (i) offer, pledge, sell, contract to sell, sell any option or contract to purchase, purchase any option or contract to sell, grant any option, right or warrant to purchase, lend, or otherwise transfer or dispose of, directly or indirectly, any shares of capital stock of the Company or any securities convertible into or exercisable or exchangeable for shares of capital stock of the Company, other than shares issued primarily as equity incentives or securities issued in transactions not primarily for capital raising; (ii) file or caused to be filed any registration statement with the SEC relating to the offering of any shares of capital stock of the Company or any securities convertible into or exercisable or exchangeable for shares of capital stock of the Company; (iii) complete any offering of debt securities of the Company, other than entering into a line of credit with a traditional bank; or (iv) enter into any swap or other arrangement that transfers to another, in whole or in part, any of the economic consequences of ownership of capital stock of the Company, whether any such transaction described in clause (i), (ii), (iii) or (iv) above is to be settled by delivery of shares of capital stock of the Company or such other securities in cash or otherwise.
Our directors, executive officers and the holders of substantially all of our equity securities have agreed, subject to certain exceptions, with the underwriters that for a period of 180 days after the date of this prospectus, they will not, except with the prior written consent of the Representative, offer, pledge, sell, contract to sell, sell any option or contract to purchase, purchase any option or contract to sell, grant any option, right or warrant to sale of or otherwise dispose of or transfer any shares of our Common Stock or any securities convertible into or exercisable or exchangeable for shares of our Common Stock, request or demand that we file a registration statement related to our Common Stock, or enter into any swap or other agreement that transfers to another, in whole or in part, directly or indirectly, the economic consequence of ownership of the Common Stock. All of our option holders and warrant holders are subject to a market stand-off agreement with us which imposes similar restrictions.
The Representative may in its sole discretion and at any time without notice release some or all of the shares subject to lock-up agreements prior to the expiration of the lock-up period. When determining whether to release shares from the lock-up agreements, the Representative will consider, among other factors, the security holder’s reasons for requesting the release, the number of shares for which the release is being requested and market conditions at the time.
Right of First Refusal
According to the terms of the underwriting agreement, the Representative shall have the right of first refusal for a period of eight (8) months after the closing of our initial public offering to participate in each and every future public and private equity and debt offerings of the Company, or any successor to or any subsidiary of the Company. The right of first refusal granted hereunder may be terminated by us for “cause,” which shall mean a material breach by the Representative of the underwriting agreement or a material failure by the Representative to provide the services as contemplated by the underwriting agreement in which case we will not be obligated to honor the right of first refusal.
| 108 |
Tail Rights
If the Company, within twelve (12) months after the termination of the engagement agreement with the Representative, effects a sale of any securities with a party introduced by the Representative, the Company shall pay to the Representative the cash discount and warrants set forth above upon the completion of such transaction, provided that such tail financing is by a party actually introduced to the Company in an offering in which the Company has direct knowledge of such party’s participation. In compliance with FINRA Rule 5110(g)(5)(B), the “tail fee” will not be payable for greater than one year and our entire underwriting agreement with the Representative is terminable if the Representative materially breaches the engagement agreement or fails to materially perform the underwriting services contemplated in the underwriting agreement. The termination of such agreement will eliminate the obligation of the Company to pay the tail fee.
Indemnification
We have agreed to indemnify the underwriters against specified liabilities, including liabilities under the Securities Act, and to contribute to payments the underwriters may be required to make in respect thereof.
Electronic Offer, Sale and Distribution of Units
A prospectus in electronic format may be made available on a website maintained by the Representative and may also be made available on a website maintained by other underwriters. The underwriters may agree to allocate a number of Units to underwriters for sale to their online brokerage account holders. Internet distributions will be allocated by the Representative to underwriters that may make internet distributions on the same basis as other allocations. In connection with the Offering, the underwriters or syndicate members may distribute prospectuses electronically. No forms of electronic prospectus other than prospectuses that are printable as Adobe® PDF will be used in connection with this Offering.
The underwriters have informed us that they do not expect to confirm sales of Units offered by this prospectus to accounts over which they exercise discretionary authority.
Other than the prospectus in electronic format, the information on any underwriter’s website and any information contained in any other website maintained by an underwriter is not part of the prospectus or the registration statement of which this prospectus forms a part, has not been approved and/or endorsed by us or any underwriter in its capacity as underwriter and should not be relied upon by investors.
| 109 |
Certain Relationships
Certain of the underwriters and their affiliates have provided and may in the future provide various investment banking, commercial banking and other financial services for us and our affiliates for which they have or may in the future receive customary fees, however, except for the right of first refusal disclosed in this prospectus, we have no present arrangements with any of the underwriters for any further services.
The Representative and certain of its affiliates are full service financial institutions engaged in, and may in the future engage in, various activities, which may include securities trading, investment banking and other commercial dealings, financial advisory, investment management, investment research, principal investment, hedging, financing and brokerage activities. The underwriters and certain of its affiliates have, from time to time, performed, and may in the future perform, various commercial and investment banking and financial advisory services for us and our affiliates, for which they received or will receive customary fees and expenses. In addition, from time to time, the underwriters and their affiliates may effect transactions for their own account or the account of customers, and hold on behalf of themselves or their customers, long or short positions in our debt or equity securities or loans, and may do so in the future.
In the ordinary course of their various business activities, the underwriters and certain of their affiliates may make or hold a broad array of investments and actively trade debt and equity securities (or related derivative securities) and financial instruments (including bank loans) for their own account and for the accounts of their customers, and such investment and securities activities may involve securities and/or instruments issued by us and our affiliates. If the underwriters or their affiliates have a lending relationship with us, they routinely hedge their credit exposure to us consistent with their customary risk management policies. The underwriters and their affiliates may hedge such exposure by entering into transactions which consist of either the purchase of credit default swaps or the creation of short positions in our securities or the securities of our affiliates, including potentially the Common Stock offered hereby. Any such short positions could adversely affect future trading prices of the Common Stock offered hereby. The underwriters and certain of their affiliates may also communicate independent investment recommendations, market color or trading ideas and/or publish or express independent research views in respect of such securities or instruments and may at any time hold, or recommend to clients that they acquire, long and/or short positions in such securities and instruments.
Selling Restrictions
Other than in the United States of America, no action has been taken by us or the underwriters that would permit a public offering of the securities offered by this prospectus in any jurisdiction where action for that purpose is required. The securities offered by this prospectus may not be offered or sold, directly or indirectly, nor may this prospectus or any other offering material or advertisements in connection with the offer and sale of any such securities be distributed or published in any jurisdiction, except under circumstances that will result in compliance with the applicable rules and regulations of that jurisdiction. Persons into whose possession this prospectus comes are advised to inform themselves about and to observe any restrictions relating to the Offering and the distribution of this prospectus. This prospectus does not constitute an offer to sell or a solicitation of an offer to buy any securities offered by this prospectus in any jurisdiction in which such an offer or a solicitation is unlawful.
Transfer Agent
The transfer agent and registrar for our Common Stock is VStock Transfer, LLC. The transfer agent and registrar’s address is 18 Lafayette Place, Woodmere, New York 11598.
| 110 |
LEGAL MATTERS
The validity of the securities offered hereby will be passed upon for us by Blank Rome LLP, New York, New York. Certain legal matters in connection with this Offering will be passed upon for the Underwriter by Carmel, Milazzo & Feil LLP, New York, New York.
EXPERTS
The consolidated financial statements of bioAffinity Technologies, Inc. at December 31, 2022, and for the year ended December 31, 2022, appearing in this prospectus and registration statement have been audited by WithumSmith+Brown, PC, independent registered public accounting firm, as set forth in their report thereon (which contains an explanatory paragraph describing conditions that raise substantial doubt about bioAffinity Technologies, Inc.’s ability to continue as a going concern as described in Note 1 to the consolidated financial statements) appearing elsewhere herein, and are included in reliance upon such report given on the authority of such firm as experts in accounting and auditing.
The financial statements of Village Oaks Pathology Services, P.A. d/b/a Precision Pathology Services (“PPLS”) at December 31, 2022, and for the year ended December 31, 2022, appearing in this prospectus and registration statement have been audited by WithumSmith+Brown, PC, independent auditors, as set forth in their report thereon (which contains an explanatory paragraph describing conditions that raise substantial doubt about PPLS’s ability to continue as a going concern as described in Note 2 to the financial statements) appearing elsewhere herein, and are included in reliance upon such report given on the authority of such firm as experts in accounting and auditing.
WHERE YOU CAN FIND ADDITIONAL INFORMATION
We have filed with the SEC a registration statement on Form S-1 relating to the securities offered by this prospectus. This prospectus, which constitutes a part of the registration statement, does not contain all of the information set forth in the registration statement. For further information regarding us and the securities offered by this prospectus, we refer you to the full registration statement, including its exhibits and schedules, filed under the Securities Act.
The SEC maintains a website at http://www.sec.gov that contains reports, information statements, and other information regarding issuers that file electronically with the SEC. Our registration statement, of which this prospectus constitutes a part, and the exhibits and schedules thereto can be downloaded from the SEC’s website. After the completion of this Offering, we will file with or furnish to the SEC periodic reports and other information. These reports and other information may be obtained from the SEC’s website as provided above.
Our website is located at https://www.bioaffinitytech.com/. Our periodic reports and other information filed with or furnished to the SEC is available, free of charge, through our website, as soon as reasonably practicable after those reports and other information are electronically filed with or furnished to the SEC. Information on our website or any other website is not incorporated by reference into this prospectus and does not constitute a part of this prospectus.
We intend to furnish or make available to our shareholders annual reports containing our audited financial statements prepared in accordance with GAAP. We also intend to furnish or make available to our shareholders quarterly reports containing our unaudited interim financial information, including the information required by Form 10-Q, for the first three fiscal quarters of each fiscal year.
DISCLOSURE
OF COMMISSION POSITION ON INDEMNIFICATION
FOR SECURITIES ACT LIABILITIES
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to our directors, officers, and controlling persons, we have been informed that in the opinion of the SEC this indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable.
| 111 |
GLOSSARY OF SELECTED TERMS
| Adenocarcinoma | A form of cancer that forms in the tissue that lines certain internal organs. Most cancers of the breast, pancreas, lung, prostate, colon, esophagus, and stomach are adenocarcinomas. | |
| Antibody | A protein that is produced by a person’s immune system to target and destroy alien substances in the blood such as bacteria or viruses. | |
| Bio-label | A tag that is chemically attached to an individual cell. These tags, or bio-labels, help to identify or track the cell on basis of its color or radioactivity depending on the type of bio-label used. | |
| Cancerization | The act of transforming a normal cell or tissue to a cancerous state. | |
| CAP | The College of American Pathologists (CAP). CAP is a professional association that, among other things, issues guidance for commercial laboratories. CAP guidance must be followed by the laboratory to receive CAP certification. CAP often works in collaboration with regulations issued by the U.S. Centers for Medicare and Medicaid under authority granted the Agency by the Clinical Laboratory Improvement Amendments (CLIA). (See also “CLIA” and “Laboratory Developed Test (LDT)”). | |
| CD320 Gene | A gene that provides instructions for making CD320 receptors. The CD320 receptor on the surface facilitates the uptake of vitamin B12, an important nutrient for human cells. Cancer cells can express large numbers of CD320 receptors on their cell surface. | |
| Cell surface receptors | Proteins that are located on the cell surface that interact, or bind, with specific molecules outside the cell called ligands. | |
| CE-marked | The letters ‘CE’ (Conformitè Europëenne) on a product signifies that products sold in the European Union have been assessed to meet high safety, health, and environmental protection requirements. | |
| CLIA |
The Clinical Laboratory Improvement Amendments of 1989 (CLIA). These amendments to U.S. law grant authority to the Centers for Medicare and Medicaid to issue regulations and guidance governing commercial laboratories. CLIA regulations are often associated with CAP guidance. (See also “Laboratory Developed Test (LDT)”).
|
|
| Cobalamin | Another name for vitamin B12. | |
| Cytology | A branch of biology that deals with the structure, function, multiplication, pathology, and life history of cells. | |
| Endocytosis | The process of actively transporting a molecule into a cell by engulfing the molecule with the cell’s membrane. | |
| Flow cytometry | A technique that can distinguish individual cells in a fluid such as blood or sputum. In the flow cytometry process, cells flow individually past a laser and this produces data to be analyzed to distinguish different cell types. Cells can be labeled to identify different types of cells. Flow cytometry has applications in fields like immunology, virology, molecular biology, cancer biology, disease diagnosis, and infectious disease monitoring. | |
| Gene expression | A biological process taking place in a cell by which the information encoded in our DNA (i.e., our genes) is converted into a product, like a protein, that can perform different cell functions. Proteins carry out most of the active functions of a cell. | |
| Gene silencing | A biological process by which an mRNA molecule is destroyed and prevented from delivering its instructions for producing a protein. | |
| Heme | The deep red, nonprotein component of hemoglobin that carries oxygen in the blood. Heme is a porphyrin. | |
| IVD | Diagnostic tests whose process of detection is performed outside the body, or in vitro. | |
| Knock-down of CD320 and LRP2 | bioAffinity uses siRNA to target and destroy the instructions encoded by the CD320 and LRP2 Genes that lead to a cessation in CD320 and LRP2 receptor production, thereby killing cancer cells with little or no harm to healthy cells. | |
| Laboratory reagent | A substance that is used in a laboratory to measure, detect, or create other substances during a chemical reaction. Reagents are the substances added to the laboratory tests to carry out a chemical reaction or to check whether any reaction occurs or not. | |
| Laboratory Developed Test (LDT) | An LDT is a type of diagnostic test that is designed, manufactured and used within a single laboratory. LDTs are performed in vitro, that is, outside the body (See also “IVD”). | |
| Low-dose computed tomography (LDCT) | A medical imaging test that uses a low-dose of radiation to create high-quality images of the inside of the human body. The radiation exposure in LDCT scans is more than a standard X-ray, but up to 90% less than a conventional CT chest scan. The only recommended screening test for lung cancer is LDCT. |
| 112 |
| LRP2 Gene | A gene that provides instructions for making the LRP2 receptor that facilitates the uptake of many proteins and some nutrients that includes vitamin B12. Cancer cells can express a large number of LRP2 receptors on their cell surface. | |
| Metabolism | The set of life-sustaining chemical reactions used by organisms to convert the energy in food to energy available for the body to stay alive, grow and reproduce, maintain the body’s structures, and respond to its environments. | |
| Negative predictive value | The probability that a patient with a negative diagnostic or screening test truly does not have the disease. Negative predictive value is a function of the incidence of a disease in a population (i.e., the estimated percentage of people who are expected to have the disease in the population) and the specificity of a test (See “Specificity”). | |
| Nodules | Abnormal tissue growths that can be found anywhere in the body. Although they are often benign, some nodules are symptoms of an underlying health condition such as cancer. | |
| Organic compound | Organic compounds are the complex compounds of carbon. These compounds can occur naturally or can be man-made (synthesized) in a laboratory. | |
| Pathology | The branch of medicine that deals with the laboratory examination of samples of body tissue for diagnostic or forensic purposes. | |
| Pivotal trial | A clinical study seeking to demonstrate the efficacy of a new diagnostic test in order to obtain approval by the U.S. FDA to market the test directly by its manufacturer. | |
| Plasma | The liquid portion of blood. Its main role is to take nutrients, hormones, and proteins to the parts of the body that need it. Cells also excrete their waste products into the plasma. | |
| Porphyrins | A class of pigments that can be either lab-produced or naturally occurring, many of which are essential to life, such as the green chlorophyll for photosynthesis in plants and the oxygen carrier, hemoglobin, that gives blood its red color. The molecular structure of all porphyrins is a large ring composed of four linked nitrogen-containing rings known as pyrroles. | |
| Positive predictive value | The probability that a patient with a positive diagnostic or screening test truly has the disease. Positive predictive value is a function of the incidence of a disease in a population (i.e., the estimated percentage of people who are expected to have the disease in the population) and the sensitivity of a test (See “Sensitivity”). |
| Pre-malignant | A term used to describe a condition that may (or is likely to) become cancer. Also called precancerous. | |
| RNA interference (RNAi) | A natural process in which small pieces of RNA shut down a cell’s ability to make certain proteins. To do so, RNAi binds to the messenger RNA (mRNA) that carries instructions for that protein. | |
| RNA | Ribonucleic acid, a naturally occurring chemical compound present in all living cells. RNA’s principal role is to act as a messenger carrying instructions from DNA for controlling the synthesis of proteins. Several types of RNA sequences are often mentioned, including: | |
| mRNA | Messenger RNA (mRNA), the molecule that carries protein-building instructions from DNA to the ribosome, the part of the cell where proteins are assembled. | |
| siRNA | Small interfering RNA (siRNA), short molecules that bind to an mRNA and target it for destruction. | |
| Sensitivity | In a diagnostic test, sensitivity is a measure of how well a test can identify true positives, meaning the test’s ability to detect a disease in a person with that disease. There is a trade-off between sensitivity and specificity, such that higher sensitivities will mean lower specificities and vice versa. | |
| Specificity | Specificity is a measure of how well a test can identify someone who does not have a disease is negative for that disease. | |
| Squamous cell carcinoma | A type of cancer that begins in squamous cells. Squamous cells are thin, flat cells that look like fish scales, and are found in the tissue that forms the surface of the skin, the lining of the hollow organs of the body, and the lining of the respiratory and digestive tracts. Most cancers of the anus, cervix, head and neck, and vagina are squamous cell carcinomas. | |
| Stage I–IV | Staging describes where cancer is located, how far the primary tumor (where the cancer started) has spread and to where, and its size. This is one method used to define how cancer is growing and advancing in the body. The lower the number, the less advanced the disease. Stage I is when cancer is relatively small and is contained where it started. Stage II is when cancer has started to spread, but is still in the early stage of disease. In Stage III, cancer has spread more so than Stage II, and may be considered a regional cancer, as opposed to local, meaning the cancer has metastasized to nearby lymph nodes, lymph vessels, or another organ. By Stage IV, cancer is advanced and has spread to multiple areas in the body. It is important to take note that each case of cancer is different, even within the same stage. | |
| Synthesis | The making of a chemical compound by combining simpler materials. Synthesis can occur both naturally and in the laboratory. | |
| Synthetic | A chemical or compound that is produced artificially in a laboratory rather than a natural system. Naturally occurring molecules can be made synthetically, and have the same molecular structure and properties as the nature-made material. | |
| TCPP | A specific synthetic (i.e., man-made) porphyrin molecule whose chemical name is meso-tetra (4-carboxyphenyl) porphine. | |
| Transfection | A laboratory technique that is used to insert foreign nucleic acid (DNA or RNA) into a cell, typically with the intention of producing a specific protein within the cell. | |
| Vitamin B12 | An essential dietary nutrient that the body needs daily in small amounts to function and stay healthy. Vitamin B12 helps make red blood cells, DNA, RNA, energy, and tissues, and keeps nerve cells healthy. It is found in liver, meat, eggs, poultry, shellfish, milk, and milk products. Chronic lack of vitamin B12 can result in anemia and central nervous system problems. |
| 113 |
TABLE OF CONTENTS
| bioAffinity Technologies, Inc. December 31, 2022 Financial Statements | |
| Report of Independent Registered Public Accounting Firm (PCAOB ID NO. 100) | F-11 |
| Consolidated Balance Sheets as of December 31, 2022 and 2021 | F-12 |
| Consolidated Statements of Operations for the years ended December 31, 2022 and 2021 | F-13 |
| Consolidated Statements of Changes in Convertible Preferred Stock and Stockholders’ Equity (Deficit) for the years ended December 31, 2022 and 2021 | F-14 |
| Consolidated Statements of Cash Flows for the years ended December 31, 2022 and 2021 | F-15 |
| Notes to Consolidated Financial Statements | F-16 |
| Village Oaks Pathology Services, P.A. d/b/a Precision Pathology Services December 31, 2022 Financial Statements | |
| INDEPENDENT AUDITOR’S REPORT | F-29 |
| Balance Sheets as of December 31, 2022 and 2021 | F-30 |
| Statements of Operations for the years ended December 31, 2022 and 2021 | F-31 |
| Statements of Stockholders’ Equity for the years ended December 31, 2022 and 2021 | F-32 |
| Statements of Cash Flows for the years ended December 31, 2022 and 2021 | F-33 |
| Notes to the Financial Statements | F-34 |
| Village Oaks Pathology Services, P.A. d/b/a Precision Pathology Services June 30, 2023 Financial Statements | |
| Balance Sheets as of June 30, 2023 (unaudited) and December 31, 2022 | F-44 |
| Unaudited Statements of Operations for the six months ended June 30, 2023 and 2022 | F-45 |
| Unaudited Statements of Stockholders’ Equity for the six months ended June 30, 2023 and 2022 | F-46 |
| Unaudited Statements of Cash Flows for the six months ended June 30, 2023 and 2022 | F-47 |
| Notes to the Unaudited Financial Statements | F-49 |
| F-1 |
bioAffinity Technologies, Inc.
Condensed Consolidated Balance sheets
| June 30, | December 31, | |||||||
| 2023 | 2022 | |||||||
| (Unaudited) | ||||||||
| ASSETS | ||||||||
| Current assets: | ||||||||
| Cash and cash equivalents | $ | $ | ||||||
| Accounts and other receivables, net | ||||||||
| Inventory | ||||||||
| Prepaid and other current assets | ||||||||
| Total current assets | ||||||||
| Property and equipment, net | ||||||||
| Other assets | ||||||||
| Total assets | $ | $ | ||||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY (DEFICIT) | ||||||||
| Current liabilities: | ||||||||
| Accounts payable | $ | $ | ||||||
| Accrued expenses | ||||||||
| Unearned revenue | ||||||||
| Loan payable | ||||||||
| Total current liabilities | ||||||||
| Total liabilities | ||||||||
| Commitments and contingencies (See Note 9) | ||||||||
| Stockholders’ equity: | ||||||||
| Preferred stock, par value $ per share; shares authorized; shares issued or outstanding at June 30, 2023, and December 31, 2022 | ||||||||
| Common stock, par value $ per share; shares authorized; issued and outstanding at June 30, 2023; and shares issued and outstanding at December 31, 2022 | ||||||||
| Additional paid-in capital | ||||||||
| Accumulated deficit | ( |
) | ( |
) | ||||
| Total stockholders’ equity | ||||||||
| Total liabilities and stockholders’ equity | $ | $ | ||||||
The accompanying notes are an integral part of these condensed consolidated financial statements.
| F-2 |
bioAffinity Technologies, Inc.
Unaudited Condensed Consolidated Statements of Operations
| Three Months Ended June 30, |
Six Months Ended June 30, |
|||||||||||||||
| 2023 | 2022 | 2023 | 2022 | |||||||||||||
| (Unaudited) | (Unaudited) | |||||||||||||||
| Revenue | $ | $ | $ | $ | ||||||||||||
| Cost of sales | ||||||||||||||||
| Gross profit | ||||||||||||||||
| Operating expenses: | ||||||||||||||||
| Research and development | ||||||||||||||||
| Clinical development | ||||||||||||||||
| General and administrative | ||||||||||||||||
| Total operating expenses | ||||||||||||||||
| Loss from operations | ( |
) | ( |
) | ( |
) | ( |
) | ||||||||
| Other income (expense): | ||||||||||||||||
| Interest income | ||||||||||||||||
| Interest expense | ( |
) | ( |
) | ( |
) | ( |
) | ||||||||
| Gain on extinguishment of debt | ||||||||||||||||
| Fair value adjustments on convertible notes payable | ||||||||||||||||
| Net loss before provision for income taxes | ( |
) | ( |
) | ( |
) | ( |
) | ||||||||
| Income tax expense | ||||||||||||||||
| Net loss | $ | ( |
) | $ | ( |
) | $ | ( |
) | $ | ( |
) | ||||
| Net loss per common share, basic and diluted | $ | ) | $ | ) | $ | ) | $ | ) | ||||||||
| Weighted average common shares outstanding, basic and diluted | ||||||||||||||||
The accompanying notes are an integral part of these unaudited condensed consolidated financial statements.
| F-3 |
bioAffinity Technologies, Inc.
Unaudited Condensed Consolidated Statements of Stockholders’ Equity (Deficit)
| For the Six Months Ended June 30, 2023 | ||||||||||||||||||||||||||||
| Preferred Stock | Common Stock | Additional Paid-in |
Accumulated | Stockholders’ | ||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Capital | Deficit | Equity | ||||||||||||||||||||||
| Balance at December 31, 2022 | $ | $ | $ | $ | ( |
) | $ | |||||||||||||||||||||
| Stock-based compensation expense | — | |||||||||||||||||||||||||||
| Net loss | — | — | ( |
) | ( |
) | ||||||||||||||||||||||
| Balance at June 30, 2023 (Unaudited) | $ | $ | $ | $ | ( |
) | $ | |||||||||||||||||||||
| For the Three Months Ended June 30, 2023 | ||||||||||||||||||||||||||||
| Preferred Stock | Common Stock | Additional Paid-in |
Accumulated | Stockholders’ | ||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Capital | Deficit | Equity | ||||||||||||||||||||||
| Balance at March 31, 2022 | $ | $ | $ | $ | ( |
) | $ | |||||||||||||||||||||
| Stock-based compensation expense | — | |||||||||||||||||||||||||||
| Net loss | — | — | ( |
) | ( |
) | ||||||||||||||||||||||
| Balance at June 30, 2023 (Unaudited) | $ | $ | $ | $ | ( |
) | $ | |||||||||||||||||||||
| For the Six Months Ended June 30, 2022 | ||||||||||||||||||||||||||||
| Convertible Preferred Stock |
Common Stock | Additional Paid-in |
Accumulated | Stockholders’ | ||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Capital | Deficit | Deficit | ||||||||||||||||||||||
| Balance at December 31, 2021 | $ | $ | $ | $ | ( |
) | $ | ( |
) | |||||||||||||||||||
| Stock-based compensation expense | — | |||||||||||||||||||||||||||
| Beneficial conversion feature for bridge notes | — | — | ||||||||||||||||||||||||||
| Debt discount for warrants issued | — | — | ||||||||||||||||||||||||||
| Net loss | — | — | ( |
) | ( |
) | ||||||||||||||||||||||
| Balance at June 30, 2022 (unaudited) | $ | $ | $ | $ | ( |
) | $ | ( |
) | |||||||||||||||||||
| For the Three Months Ended June 30, 2022 | ||||||||||||||||||||||||||||
| Convertible Preferred Stock |
Common Stock | Additional Paid-in |
Accumulated | Stockholders’ | ||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Capital | Deficit | Deficit | ||||||||||||||||||||||
| Balance at March 31, 2022 | $ | $ | $ | $ | ( |
) | $ | ( |
) | |||||||||||||||||||
| Stock-based compensation expense | — | |||||||||||||||||||||||||||
| Debt discount for warrants issued | — | — | ( |
) | ( |
) | ||||||||||||||||||||||
| Net loss | — | — | ( |
) | ( |
) | ||||||||||||||||||||||
| Balance at June 30, 2022 (unaudited) | $ | $ | $ | $ | ( |
) | $ | ( |
) | |||||||||||||||||||
The accompanying notes are an integral part of these condensed consolidated financial statements.
| F-4 |
bioAffinity Technologies, Inc.
Unaudited Condensed Consolidated Statements of Cash Flows
| Six Months Ended June 30, 2023 | ||||||||
| 2023 | 2022 | |||||||
| Cash flows from operating activities | ||||||||
| Net loss | $ | ( |
) | $ | ( |
) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Depreciation and amortization | ||||||||
| Accretion of debt issuance costs | ||||||||
| Fair value adjustments on convertible notes payable | ( |
) | ||||||
| Stock-based compensation expense | ||||||||
| Gain on extinguishment of debt | ( |
) | ||||||
| Changes in operating assets and liabilities: | ||||||||
| Accounts and other receivables | ( |
) | ( |
) | ||||
| Inventory | ( |
) | ( |
) | ||||
| Prepaid expenses and other assets | ( |
) | ||||||
| Accounts payable | ( |
) | ||||||
| Accrued expenses | ( |
) | ( |
) | ||||
| Accrued interest | ||||||||
| Unearned revenue | ||||||||
| Net cash used in operating activities | ( |
) | ( |
) | ||||
| Cash flows from investing activities | ||||||||
| Purchase of equipment | ( |
) | ||||||
| Net cash used in investing activities | ( |
) | ||||||
| Cash flows from financing activities | ||||||||
| Payment on loan payable | ( |
) | ||||||
| Payment of debt | ( |
) | ||||||
| Issuance of loan payable | ||||||||
| Proceeds from issuance of convertible notes payable | ||||||||
| Payment of deferred offering costs | ( |
) | ||||||
| Payment of debt issuance costs | ( |
) | ||||||
| Net cash used in financing activities | ( |
) | ( |
) | ||||
| Net decrease in cash and cash equivalents | ( |
) | ( |
) | ||||
| Cash and cash equivalents at beginning of period | ||||||||
| Cash and cash equivalents at end of period | $ | $ | ||||||
| Supplemental disclosures of cash flow information: | ||||||||
| Income taxes paid in cash | $ | $ | ||||||
| Interest expense paid in cash | $ | $ | ||||||
| Noncash financing activities: | ||||||||
| Fair value of warrants issued to placement agents | $ | $ | ||||||
| Beneficial conversion feature for bridge notes | $ | $ | ||||||
The accompanying notes are an integral part of these condensed consolidated financial statements.
| F-5 |
bioAffinity Technologies, Inc.
Notes To Condensed Consolidated Financial Statements
(unaudited)
Note 1. NATURE OF OPERATIONS, ORGANIZATION, AND BASIS OF PRESENTATION
Description of Business
bioAffinity Technologies, Inc., a Delaware corporation (the “Company,” “we,” “our” or “bioAffinity Technologies”), addresses the need for noninvasive diagnosis of early-stage cancer and diseases of the lung. The Company also is conducting early-stage research focused on advancing therapeutic discoveries that could result in broad-spectrum cancer treatments. bioAffinity Technologies develops proprietary noninvasive diagnostic tests using technology that preferentially target cancer cells and cell populations indicative of a diseased state. Our first diagnostic test, CyPath® Lung, is a noninvasive test for early detection of lung cancer, the leading cause of cancer-related deaths. Research and optimization of our proprietary platform for in vitro diagnostics and technologies are conducted in laboratories at The University of Texas at San Antonio. We are developing our platform technologies so that in the future, they will be able to detect, monitor, and treat diseases of the lung and other cancers.
Organization
The Company was formed on March 26, 2014, as a Delaware corporation with its corporate offices located in San Antonio, Texas. On June 15, 2016, the Company formed a wholly owned subsidiary, OncoSelect® Therapeutics, LLC, as a Delaware limited liability company.
Basis of Presentation
The accompanying unaudited condensed consolidated financial statements have been prepared in accordance with generally accepted accounting principles in the United States (“GAAP”) and pursuant to the rules and regulations of the SEC for interim financial reporting. The condensed consolidated financial statements are unaudited and in management’s opinion include all adjustments, including normal recurring adjustments and accruals, necessary for a fair presentation of the results for the interim periods presented. Operating results for the periods presented are not necessarily indicative of the results that may be expected for the fiscal year ended December 31, 2023, or any future period. These unaudited condensed consolidated financial statements should be read in conjunction with the audited annual consolidated financial statements and notes included in the 2022 Form 10-K filed with the SEC.
Liquidity and Capital Resources
In accordance with Accounting Standards Update (“ASU”) 2014-15, Presentation of Financial Statements – Going Concern (Subtopic 205-40), the Company has evaluated whether there are conditions and events that raise substantial doubt about the Company’s ability to continue as a going concern for at least one year after the date the condensed consolidated financial statements are issued.
The
Company has incurred significant losses and negative cash flows from operations since inception and expects to continue to incur losses
and negative cash flows for the foreseeable future. As a result, the Company had an accumulated deficit of $
COVID-19
The rapid global spread of the COVID-19 virus since December 2019 has affected production and sales worldwide, disrupted supply chains across a range of industries, and created significant economic volatility. The impact of COVID-19 on the Company’s operational and financial performance will depend on numerous factors, including the spread, duration, and intensity of the pandemic (including resurgences), the emergence of new viral variants, and the impact of the pandemic on the Company’s customers, employees, clinical trial sites, and vendors.
| F-6 |
As the COVID-19 pandemic continues to evolve, the ultimate impact on the Company’s operations is highly uncertain and subject to change and will depend on future developments, which cannot be accurately predicted, including the duration of the pandemic, additional or modified government actions, and the actions taken to contain COVID-19 or address its impact, among others. Management does not yet know the full extent of potential delays or impacts on the Company, clinical trials, research programs, healthcare systems, or the global economy but continues to monitor the situation closely.
Note 2. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Use of Estimates
The preparation of condensed consolidated financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates. Significant estimates include the valuation allowance on the Company’s deferred tax assets, and the useful lives of fixed assets.
Principles of Consolidation
The accompanying condensed consolidated financial statements include all of the accounts of the Company and its wholly owned subsidiary, OncoSelect® Therapeutics, LLC. All significant intercompany balances and transactions have been eliminated.
Cash and Cash Equivalents
For the purpose of the statement of cash flows, the Company considers all highly liquid investments with original maturities of three months or less at the time of purchase to be cash equivalents. Cash equivalents are stated at cost, which approximates market value, because of the short maturity of these instruments.
Concentration of Risk
The
Company has significant cash balances at financial institutions which throughout the year regularly exceed the federally insured limit
of $
Advertising expense
The
Company expenses all advertising costs as incurred. Advertising expense was approximately $
Basic earnings (loss) per share is computed by dividing net income (loss) attributable to common stockholders by the weighted-average number of shares of the Company’s common stock, par value $0.007 per share (the “Common Stock”) outstanding during the period. Diluted earnings per share is computed by dividing net income attributable to common stockholders by the sum of the weighted-average number of shares of Common Stock outstanding during the period and the weighted-average number of dilutive Common Stock equivalents outstanding during the period, using the treasury stock method. Dilutive Common Stock equivalents are comprised of in-the-money stock options, convertible notes payable, and warrants based on the average stock price for each period using the treasury stock method.
| As of June 30, | ||||||||
| 2023 | 2022 | |||||||
| Convertible preferred stock | ||||||||
| Shares underlying options outstanding | ||||||||
| Shares underlying warrants outstanding | ||||||||
| Shares underlying convertible notes | ||||||||
| F-7 |
Revenue Recognition
Revenue
is generated in three ways for the six months and three months, respectively, ended June 30, 2023: (1) royalties from the Company’s
diagnostic test, CyPath® Lung for approximately $
To determine revenue recognition for the arrangements that the Company determines are within the scope of ASC 606, Revenue from Contracts with Customers, the Company performs the following five steps: (1) identify the contract(s) with a customer, (2) identify the performance obligations in the contract, (3) determine the transaction price, (4) allocate the transaction price to the performance obligations in the contract, and (5) recognize revenue when (or as) the entity satisfies a performance obligation.
Reclassifications
Certain
prior year balances have been reclassified to conform to current year presentation. The Company reclassified patent expenses and annuity
costs of approximately $
Recent Accounting Pronouncements
The Company continues to monitor new accounting pronouncements issued by the Financial Accounting Standards Board (“FASB”) and does not believe any accounting pronouncements issued through the date of this Quarterly Report will have a material impact on the Company’s condensed consolidated financial statements.
Note 3. PREPAID EXPENSES AND OTHER CURRENT ASSETS
Prepaid expenses and other current assets are summarized below:
| June 30, 2023 | December 31, 2022 | |||||||
| Prepaid insurance | $ | $ | ||||||
| Legal and professional | ||||||||
| Other | ||||||||
| Total prepaid expenses and other current assets | $ | $ | ||||||
Note 4. PROPERTY AND EQUIPMENT, NET
Property and equipment are summarized below:
| June 30, 2023 | December 31, 2022 | |||||||
| Lab equipment | $ | $ | ||||||
| Computers and software | ||||||||
| Accumulated depreciation | ( |
) | ( |
) | ||||
| Total property and equipment, net | $ | $ | ||||||
Depreciation
expense was approximately $
Note 5. ACCRUED EXPENSES
Accrued expenses are summarized below:
| June 30, 2023 | December 31, 2022 | |||||||
| Compensation | $ | $ | ||||||
| Legal and professional | ||||||||
| Clinical | ||||||||
| Other | ||||||||
| Total accrued expenses | $ | $ | ||||||
| F-8 |
Note 6. UNEARNED REVENUE
During
the three months ended June 30, 2023, the Company engaged in an observational study of CyPath® Lung with the DOD. A total
of 70 CyPath® Lung units were ordered and shipped. However, in compliance with FASB ASC 606, the performance obligation was complete
for only 13 units as of June 30, 2023. The performance obligation is deemed complete after samples have been collected and processed
and results analyzed. The unearned revenue balance amounted to approximately $
Note 7. LOAN PAYABLE
In
September 2022, the Company obtained short-term financing of approximately $
Note 8. FAIR VALUE MEASUREMENTS
The Company analyzes all financial instruments with features of both liabilities and equity under the FASB accounting standard for such instruments. Under this standard, financial assets and liabilities are classified in their entirety based on the lowest level of input that is significant to the fair value measurement.
The estimated fair value of certain financial instruments, including cash and cash equivalents, accounts and other receivables, prepaid and other current assets, accounts payable, accrued expenses, and loan payable, are carried at historical cost basis, which approximates their fair values because of the short-term nature of these instruments.
Note 9. COMMITMENTS AND CONTINGENCIES
Operating Leases
The
Company leases its corporate offices under a month-to-month agreement and leases its laboratory and additional office space under an
operating lease that is renewable annually by written notice by the Company and will require renewal in February 2024. Rent expense for
office and lab space amounted to approximately $
Legal Matters
From time to time, the Company is involved in various disputes and litigation matters that arise in the ordinary course of business. To date, the Company has no material pending legal proceedings.
Note 10. COMMON STOCK
Common Stock
The Company has authorized a total of shares of Common Stock, $ par value per share. On June 6, 2023, the Company received stockholder approval to increase the number of authorized shares from shares to shares. The Company has issued shares of Common Stock as of June 30, 2023, and shares of Common Stock as of December 31, 2022.
The Company grants options and restricted stock awards under its 2014 Equity Incentive Plan (the “Plan”). Under the Plan, the Company is authorized to grant options or restricted stock for up to million shares of Common Stock. On June 6, 2023, the Company received stockholder approval to increase the number of authorized shares from to . Options or restricted stock awards may be granted to employees, the Company’s board of directors, and external consultants who provide services to the Company. Options and restricted stock awards granted under the Plan have vesting schedules with terms of one to three years and become fully exercisable based on specific terms imposed at the date of grant. The Plan will terminate according to the respective terms of the Plan in September 2026.
| Three Months Ended June 30, |
Six Months Ended June 30, |
|||||||||||||||
| 2023 | 2022 | 2023 | 2022 | |||||||||||||
| Research and development | $ | $ | $ | $ | ||||||||||||
| General and administrative | ||||||||||||||||
| $ | $ | $ | $ | |||||||||||||
| F-9 |
| Number of options |
Weighted-average exercise price |
Weighted-average remaining contractual term (in years) |
Aggregate intrinsic value |
|||||||||||||
| Outstanding at December 31, 2022 | $ | |||||||||||||||
| Granted | ||||||||||||||||
| Exercised | ||||||||||||||||
| Forfeited | ||||||||||||||||
| Outstanding at June 30, 2023 | $ | $ | ||||||||||||||
| Vested and exercisable at June 30, 2023 | $ | $ | ||||||||||||||
As of June 30, 2023, there was unrecognized compensation cost related to non-vested stock options. During the six months ended June 30, 2023 and 2022, options were exercised. During the six months ended June 30, 2023, options were issued by the Company to purchase shares of Common Stock. During the six months ended June 30, 2022, the Company issued options to purchase shares of Common Stock. The per share weighted-average fair value of the options granted during 2022 was estimated at $ on the date of grant.
| Number of restricted stock awards (RSA) | Weighted-average grant price | FMV on grant date | Vested number of RSA | Unvested number of RSA | ||||||||||||||||
| Balance at December 31, 2022 | $ | $ | ||||||||||||||||||
| Granted | ||||||||||||||||||||
| Forfeited | ||||||||||||||||||||
| Balance at June 30, 2023 | $ | $ | ||||||||||||||||||
During the six months ended June 30, 2023, the Company issued restricted stock awards (RSAs) for shares of Common Stock to employees, non-employees, and the board of directors. The shares vest in equal monthly installments over terms of between immediately up to three years, subject to the employees and non-employees providing continuous service through the vesting date. During the six months ended June 30, 2023, shares vested from RSAs granted prior to January 1, 2023, and shares vested from RSAs granted during the six months ended June 30, 2023.
During the six months ended June 30, 2022, the Company issued RSAs for shares of Common Stock to employees and non-employees. The shares vest in equal monthly installments over terms of between immediately up to one year, subject to the employees and non-employees providing continuous service through the vesting date. During the six months ended June 30, 2022, approximately shares vested from RSAs previously issued.
| 2022 | ||||
| Fair value of Common Stock | $ | |||
| Volatility | % | |||
| Expected term (years) | ||||
| Risk-free interest rate | % | |||
| Dividend yield | % | |||
Note 12. WARRANTS
The Company accounts for Common Stock warrants as either equity instruments or derivative liabilities depending on the specific terms of the warrant agreement. Warrants are accounted for as derivative liabilities if the warrants allow for cash settlement or provide for modification of the warrant exercise price in the event subsequent sales of Common Stock by the Company are at a lower price per share than the then-current warrant exercise price. The Company classifies derivative warrant liabilities on the condensed consolidated balance sheet at fair value, and changes in fair value during the periods presented in the condensed consolidated statement of operations, which is revalued at each balance sheet date subsequent to the initial issuance of the stock warrant.
As
of June 30, 2023, and December 31, 2022, the Company had
Note 13. SUBSEQUENT EVENTS
The Company evaluated subsequent events and transactions that occurred after the balance sheet date up to the date that the condensed consolidated financial statements were available to be issued. Based upon this review, the Company did not identify any subsequent events that would have required adjustment or disclosure in the condensed consolidated financial statements.
| F-10 |
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders of
bioAffinity Technologies, Inc.
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated balance sheets of bioAffinity Technologies, Inc. (the “Company”) as of December 31, 2022 and 2021, the related consolidated statements of operations, changes in convertible preferred stock and stockholders’ equity (deficit), and cash flows, for each of the two years in the period ended December 31, 2022, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the consolidated financial position of the Company as of December 31, 2022 and 2021 and the consolidated results of its operations and its cash flows for each of the two years in the period ended December 31, 2022, in conformity with accounting principles generally accepted in the United States of America.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB and in accordance with auditing standards generally accepted in the United States of America. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ WithumSmith+Brown, PC
We have served as the Company’s auditor since 2021.
New York, New York
March 31, 2023
PCAOB ID Number 100
| F-11 |
bioAffinity Technologies, Inc.
consolidated Balance Sheets
| December 31, | ||||||||
| 2022 | 2021 | |||||||
| ASSETS | ||||||||
| Current assets: | ||||||||
| Cash and cash equivalents | $ | $ | ||||||
| Accounts and other receivables, net | ||||||||
| Inventory | ||||||||
| Prepaid expenses and other current assets | ||||||||
| Total current assets | ||||||||
| Deferred offering costs | ||||||||
| Property and equipment, net | ||||||||
| Other assets | ||||||||
| Total assets | $ | $ | ||||||
| LIABILITIES, CONVERTIBLE PREFERRED STOCK, AND STOCKHOLDERS’ EQUITY (DEFICIT) | ||||||||
| Current liabilities: | ||||||||
| Accounts payable | $ | $ | ||||||
| Accrued expenses | ||||||||
| Accrued interest | ||||||||
| Current portion of Paycheck Protection Program loan | ||||||||
| Loan payable | ||||||||
| Convertible notes payable at fair value | ||||||||
| Total current liabilities | ||||||||
| Paycheck Protection Program loan, less current portion | ||||||||
| Total liabilities | ||||||||
| Commitments and contingencies (See Note 9) | ||||||||
| Convertible preferred stock, par value $ per share; shares authorized; and shares issued and outstanding, aggregate liquidation preference of $ |
||||||||
| Stockholders’ equity (deficit): | ||||||||
| Preferred stock, shares issued or outstanding at December 31, 2022 and 2021, respectively | ||||||||
| Common Stock, par value $ per share; shares authorized; and shares issued and outstanding as of December 31, 2022 and 2021, respectively | ||||||||
| Additional paid-in capital | ||||||||
| Accumulated deficit | ( |
) | ( |
) | ||||
| Total stockholders’ equity (deficit) | ( |
) | ||||||
| Total liabilities, convertible preferred stock, and stockholders’ equity (deficit) | $ | $ | ||||||
The accompanying notes are an integral part of these consolidated financial statements.
| F-12 |
bioAffinity Technologies, Inc.
consolidated Statements of Operations
For the Years Ended December 31, 2022 and 2021
| 2022 | 2021 | |||||||
| Revenue | $ | $ | ||||||
| Cost of sales | ||||||||
| Gross profit | ||||||||
| Operating expenses: | ||||||||
| Research and development | ||||||||
| Clinical development | ||||||||
| Selling, general and administrative | ||||||||
| Total operating expenses | ||||||||
| Loss from operations | ( |
) | ( |
) | ||||
| Other income (expense): | ||||||||
| Interest income | ||||||||
| Interest expense | ( |
) | ( |
) | ||||
| Gain on extinguishment of debt | ||||||||
| Fair value of warrants issued | ( |
) | ||||||
| Fair value adjustments on convertible notes payable | ( |
) | ||||||
| Loss before income taxes | ( |
) | ( |
) | ||||
| Income tax expense | ( |
) | ( |
) | ||||
| Net loss | $ | ( |
) | $ | ( |
) | ||
| Net loss per common share, basic and diluted | $ | ) | $ | ) | ||||
| Weighted average common shares outstanding | ||||||||
The accompanying notes are an integral part of these consolidated financial statements.
| F-13 |
bioAffinity Technologies, Inc.
consolidated Statements of Changes in Convertible preferred stock and Stockholders’ Equity (Deficit)
For the Years Ended December 31, 2022 and 2021
| Convertible | Additional | Stockholders’ | ||||||||||||||||||||||||||
| Preferred Stock | Common Stock | Paid-in | Accumulated | Equity | ||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Capital | Deficit | (Deficit) | ||||||||||||||||||||||
| Balance at December 31, 2020 | $ | $ | $ | $ | ( |
) | $ | ( |
) | |||||||||||||||||||
| Stock-based compensation expense | — | |||||||||||||||||||||||||||
| Fair value of warrants issued | — | — | ||||||||||||||||||||||||||
| Beneficial conversion feature for bridge notes | — | — | ||||||||||||||||||||||||||
| Debt discount for warrants issued | — | — | ||||||||||||||||||||||||||
| Net loss | — | — | ( |
) | ( |
) | ||||||||||||||||||||||
| Balance at December 31, 2021 | $ | $ | $ | $ | ( |
) | $ | ( |
) | |||||||||||||||||||
| Stock-based compensation expense | — | |||||||||||||||||||||||||||
| Beneficial conversion feature for bridge notes | — | — | ||||||||||||||||||||||||||
| Return of capital from stock split | — | — | ( |
) | ( |
) | ||||||||||||||||||||||
| Debt discount for warrants issued | — | — | ||||||||||||||||||||||||||
| Common stock issued upon initial public offering, net of underwriters’ commission and offering costs of $ |
— | |||||||||||||||||||||||||||
| Common stock issued on conversion of convertible preferred stock | ( |
) | $ | ( |
) | |||||||||||||||||||||||
| Common stock issued on conversion of notes payable | — | |||||||||||||||||||||||||||
| Exercise of warrants | — | |||||||||||||||||||||||||||
| Exercise of stock options | — | |||||||||||||||||||||||||||
| Net loss | — | — | ( |
) | ( |
) | ||||||||||||||||||||||
| Balance at December 31, 2022 | $ | $ | $ | $ | ( |
) | $ | |||||||||||||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
| F-14 |
bioAffinity Technologies, Inc.
consolidated Statements of Cash Flows
For the Years Ended December 31, 2022 and 2021
| 2022 | 2021 | |||||||
| Cash flows from operating activities | ||||||||
| Net loss | $ | ( |
) | $ | ( |
) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Depreciation and amortization | ||||||||
| Accretion of debt issuance costs | ||||||||
| Fair value adjustments on convertible notes payable | ( |
) | ||||||
| Stock-based compensation expense | ||||||||
| Fair value of warrants issued | ||||||||
| Gain on extinguishment of debt | ( |
) | ( |
) | ||||
| Changes in operating assets and liabilities: | ||||||||
| Accounts and other receivables | ( |
) | ||||||
| Inventory | ( |
) | ||||||
| Prepaid expenses and other assets | ( |
) | ( |
) | ||||
| Accounts payable | ||||||||
| Accrued expenses | ||||||||
| Accrued interest | ||||||||
| Net cash used in operating activities | ( |
) | ( |
) | ||||
| Cash flows from investing activities | ||||||||
| Purchase of property and equipment | ( |
) | ||||||
| Net cash used in investing activities | ( |
) | ||||||
| Cash flows from financing activities | ||||||||
| Proceeds from loan payable | ||||||||
| Payment on loans payable | ( |
) | ||||||
| Proceeds from issuance of convertible notes payable | ||||||||
| Repayment of convertible loan payable | ( |
) | ||||||
| Proceeds from issuance of common stock from the initial public offering, net of underwriting discounts, commissions and offering expenses of approximately $ |
||||||||
| Exercise of warrants | ||||||||
| Exercise of stock options | ||||||||
| Return of capital from stock split | ( |
) | ||||||
| Payment of debt issuance costs | ( |
) | ( |
) | ||||
| Net cash provided by financing activities | ||||||||
| Net increase in cash and cash equivalents | ||||||||
| Cash and cash equivalents at beginning of year | ||||||||
| Cash and cash equivalents at end of year | $ | $ | ||||||
| Supplemental disclosures of cash flow information: | ||||||||
| Income taxes paid in cash | $ | $ | ||||||
| Interest paid | $ | |||||||
| Conversion of convertible preferred stock into common stock | $ | |||||||
| Conversion of convertible notes payable into common stock | $ | |||||||
| Fair value of warrants issued to placement agents | $ | $ | ||||||
| Beneficial conversion feature for bridge notes | $ | $ | ||||||
The accompanying notes are an integral part of these consolidated financial statements.
| F-15 |
bioAffinity Technologies, Inc.
Notes to Consolidated Financial Statements
For the Years Ended December 31, 2022 and 2021
Note 1. BASIS OF PRESENTATION, ORGANIZATION AND NATURE OF OPERATIONS
Description of Business
bioAffinity Technologies, Inc., a Delaware corporation (the “Company,” “we,” or “our”), addresses the need for noninvasive diagnosis of early-stage cancer and diseases of the lung and for targeted cancer treatment. The Company develops proprietary noninvasive diagnostic tests and cancer therapeutics using technology that preferentially targets cancer cells and cell populations indicative of a diseased state. Our first diagnostic test, CyPath® Lung, is a noninvasive test for early detection of lung cancer, the leading cause of cancer-related deaths. Research and optimization of our proprietary platform for in vitro diagnostics and technologies are conducted in our laboratories at The University of Texas at San Antonio. We are developing our platform technologies so that, in the future, they will be able to detect, monitor, and treat diseases of the lung and other cancers.
Organization and Initial Public Offering
The Company was formed on March 26, 2014, as a Delaware corporation with its corporate offices located in San Antonio, Texas. On June 15, 2016, the Company formed a wholly owned subsidiary, OncoSelect® Therapeutics, LLC, as a Delaware limited liability company.
On
September 6, 2022, the Company completed its initial public offering (the “IPO”) of
units (the “Units”) at an offering price of $
per Unit (the “Offering Price”).
In
connection with the closing of the IPO, the Company converted
shares of the convertible preferred stock into
shares of Common Stock. Additionally, the Company converted approximately $
In
June 2022, the Company completed a
Basis of Presentation
The consolidated financial statements of the Company have been prepared in accordance with U.S. accounting principles generally accepted (“GAAP”).
In accordance with Accounting Standards Update (“ASU”) 2014-15, Disclosure of Uncertainties About an Entity’s Ability to Continue as a Going Concern (Subtopic 205-40), the Company has evaluated whether there are conditions and events that raise substantial doubt about the Company’s ability to continue as a going concern for at least one year after the date the consolidated financial statements are issued.
The
Company has incurred significant losses and negative cash flows from operations since inception and expects to continue to incur losses
and negative cash flows for the foreseeable future. As a result, the Company had an accumulated deficit of $
COVID-19
The rapid global spread of the COVID-19 virus since December 2019 has affected production and sales, and disrupted supply chains across a range of industries. The impact of COVID-19 on the Company’s operations and financial performance will depend on numerous factors, including but not limited to the duration and spread of the virus and the impact on the Company’s customers, employees, clinical trial sites, and vendors.
| F-16 |
bioAffinity Technologies, Inc.
Notes to Consolidated Financial Statements
For the Years Ended December 31, 2022 and 2021
As the COVID-19 pandemic continues to evolve, the ultimate impact of the pandemic on the Company’s operations is highly uncertain and subject to change and will depend on future developments, which cannot be accurately predicted, including the duration of the pandemic, additional or modified government actions, and the actions taken to contain COVID-19 or address its impact, among others. Management does not yet know the full extent of potential delays or impacts on the Company, clinical trials, research programs, healthcare systems, or the global economy, but continues to monitor the situation closely.
Note 2. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Use of Estimates
The preparation of consolidated financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates. Significant estimates include: the fair value of the Company’s Common Stock used to measure stock-based compensation for options granted to employees and nonemployees; the valuation allowance on the Company’s deferred tax assets; and the fair value of the convertible notes payable.
Principles of Consolidation
The accompanying consolidated financial statements include all of the accounts of the Company and its wholly owned subsidiary, Oncoselect Therapeutics, LLC. All significant intercompany balances and transactions have been eliminated in consolidation.
Cash and Cash Equivalents
For the purpose of the statement of cash flows, the Company considers all highly liquid investments with original maturities of three months or less at the time of purchase to be cash equivalents. Cash equivalents are stated at cost, which approximates market value, because of the short maturity of these instruments.
Concentration of Risk
The Company has significant cash balances at financial institutions which
throughout the year regularly exceed the federally insured limit of $
Accounts and Other Receivables, Net
Accounts and other receivables, net consists of amounts invoiced to Precision Pathology Services (“Precision Pathology”), a CAP-accredited, CLIA-certified clinical pathology laboratory and our licensee for royalties from sales of our first diagnostic test, CyPath® Lung.
The allowance for doubtful accounts is based on forecasted losses and a review on a specific identification basis of the collectability of outstanding receivables. As of December 31, 2022 and 2021, there is no allowance for doubtful accounts.
Prepaid Expenses and Other Assets
Prepaid expenses and other assets consist of prepaid insurance, maintenance contracts, dues, and legal retainers, etc. Expense is calculated using the straight-line method over the estimated useful lives of the respective term of service.
Deferred Offering Costs
The
Company capitalizes certain legal, accounting, and other third-party fees that are directly related to the Company’s equity financings,
including its IPO, until such financings are consummated. After consummation of the equity financing, these costs are recorded as a reduction
of the proceeds received as a result of the financing. The Company capitalized certain legal, accounting, and other third-party fees
that were directly related to the Company’s IPO. After the completion of the IPO in September 2022, total deferred offering costs
of approximately $
Property and Equipment, Net
Property
and equipment are stated at cost less accumulated depreciation and amortization. Depreciation is calculated using the straight-line method
over the estimated useful lives of the respective assets, generally three (
Property and equipment is reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of such assets may not be recoverable. The Company recognizes an impairment charge in the event the net book value of such assets exceeds the future undiscounted cash flows attributable to the asset group. No impairment losses were incurred during the years ended December 31, 2022 and 2021, respectively.
| F-17 |
bioAffinity Technologies, Inc.
Notes to Consolidated Financial Statements
For the Years Ended December 31, 2022 and 2021
Patent Expenses
Costs related to filing and pursuing patent applications, as well as costs related to maintaining the Company’s existing patent portfolio, are recorded as expenses as incurred since recoverability of such expenditures is uncertain.
Compensation expense related to stock options granted to employees and non-employees is measured at the grant date based on the estimated fair value of the award and is recognized on a straight-line basis over the requisite service period. Forfeitures are recognized as a reduction of stock-based compensation expense as they occur. The Company estimates the fair value of stock option grants using the Black-Scholes option pricing model.
The Black-Scholes option pricing model used to compute share-based compensation expense requires use of accounting judgment and financial estimates. Items requiring estimation include the expected term option holders will retain their vested stock options before exercising them and the estimated volatility of the Company’s Common Stock price over the expected term of a stock option. Application of alternative assumptions could result in different share-based compensation amounts being recorded in the financial statements. See Note 11 for additional disclosures related to stock-based compensation.
Advertising expense
The Company expenses all advertising costs as incurred. Advertising expense
was approximately $
Income Taxes
Income taxes are accounted for under the asset and liability method. Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases and operating loss and tax credit carryforwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in operations in the period that includes the enactment date. A valuation allowance is provided when it is more likely than not that some portion or all of a deferred tax asset will not be realized. The ultimate realization of deferred tax assets is dependent upon the generation of future taxable income and the reversal of deferred tax liabilities during the period in which the related temporary difference becomes deductible. The Company includes interest and penalties related to uncertain tax positions as part of income tax expense, if any. No such interest or penalties were recognized during the years ended December 31, 2022 and 2021, and the Company had no accruals for interest and penalties at December 31, 2022 or 2021.
Revenue Recognition
Our revenue is generated exclusively from royalties for our first diagnostic test, CyPath® Lung, from sales by Precision Pathology that began a limited market launch in the second quarter of 2022 to pulmonologists in the San Antonio, Texas, area designed to refine future positioning and develop strategic insight for our CyPath® Lung test. The services are completed upon release of a patient’s test result to the ordering healthcare provider.
To determine revenue recognition for the arrangements that the Company determines are within the scope of ASC 606, Revenue from Contracts with Customers, the Company performs the following five steps: (1) identify the contract(s) with a customer, (2) identify the performance obligations in the contract, (3) determine the transaction price, (4) allocate the transaction price to the performance obligations in the contract, and (5) recognize revenue when (or as) the entity satisfies a performance obligation.
Basic earnings (loss) per share is computed by dividing net income (loss) attributable to Common stockholders by the weighted-average number of common shares outstanding during the period. Diluted earnings per share is computed by dividing net income attributable to Common stockholders by the sum of the weighted-average number of common shares outstanding during the period and the weighted-average number of dilutive common share equivalents outstanding during the period, using the treasury stock method. Dilutive common share equivalents are comprised of in-the-money stock options, convertible notes payable, and warrants, based on the average stock price for each period using the treasury stock method. The following potentially dilutive securities have been excluded from the computations of weighted average shares outstanding as of December 31, 2022 and 2021, as they would be anti-dilutive:
| Year Ended December 31, | ||||||||
| 2022 | 2021 | |||||||
| Convertible preferred stock | ||||||||
| Shares underlying options outstanding | ||||||||
| Shares underlying warrants outstanding | ||||||||
| Shares underlying convertible notes outstanding | ||||||||
| F-18 |
bioAffinity Technologies, Inc.
Notes to Consolidated Financial Statements
For the Years Ended December 31, 2022 and 2021
Segment Information
The Company is organized as a single operating segment, whereby its chief operating decision maker assesses the performance of and allocates resources to the business as a whole.
Fair Value of Financial Instruments
Assets and liabilities recorded at fair value on a recurring basis in the consolidated balance sheets are categorized based upon the level of judgment associated with the inputs used to measure their fair values. Fair value is defined as the exchange price that would be received for an asset or an exit price that would be paid to transfer a liability in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. Valuation techniques used to measure fair value must maximize the use of observable inputs and minimize the use of unobservable inputs.
The three-tier fair value hierarchy for disclosure of fair value measurements is as follows:
| ● | Level 1 inputs consist of unadjusted quoted prices in active markets for identical assets or liabilities and have the highest priority. | |
| ● | Level 2 valuations are based on quoted prices in markets that are not active. | |
| ● | Level 3 valuations are based on inputs that are unobservable and supported by little or no market activity. |
See Note 7 for the fair value hierarchy table and inputs used in the fair value measurement for assets and liabilities.
Research and Development
Research and development costs are charged to expense as incurred. The Company’s research and development expenses consist primarily of expenditures for lab operations, preclinical studies, compensation, and consulting costs.
The
Company incurred research and development expenses of $
Accrued Research and Development Costs
The Company records accrued liabilities for estimated costs of research and development activities conducted by service providers, which include preclinical studies. The Company records the estimated costs of research and development activities based upon the estimated amount of services provided but not yet invoiced and includes these costs in accrued expenses in the accompanying balance sheets and within research and development expense in the accompanying consolidated statements of operations.
The Company accrues for these costs based on factors such as estimates of the work completed and in accordance with agreements established with service providers. The Company makes significant judgments and estimates in determining the accrued expenses balance in each reporting period. As actual costs become known, the Company adjusts its accrued liabilities. The Company has not experienced any material differences between accrued costs and actual costs incurred since its inception.
Regulatory Matters
Regulations imposed by federal, state, and local authorities in the United States are a significant factor in providing medical care. In the United States, drugs, biological products, and medical devices are regulated by the United States Food, Drug and Cosmetic Act, which is administered by the U.S. Food and Drug Administration (“FDA”) and the Center for Medicare and Medicaid. The Company has not yet obtained marketing authorization from the FDA but is able to market its CyPath® Lung test as a Laboratory Developed test licensed to and sold by Precision Pathology Services, a CAP-accredited, CLIA-certified clinical pathology laboratory.
Reclassifications
Certain
prior year balances have been reclassified to conform to current year presentation. The Company reclassified patent and annuity costs of approximately $
| F-19 |
bioAffinity Technologies, Inc.
Notes to Consolidated Financial Statements
For the Years Ended December 31, 2022 and 2021
Recently Issued Accounting Pronouncements
In December 2019, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) No. 2019-12, Income Taxes (Topic 740): Simplifying the Accounting for Income Taxes (ASU 2019-12). ASU 2019-12 removes certain exceptions to the general principles in Topic 740 and also clarifies and amends existing guidance to improve consistency in application. ASU 2019-12 will be effective for public entities for interim and annual periods beginning after December 15, 2020, with early adoption permitted. The Company adopted ASU 2019-12 and concluded there is no impact on the Company’s consolidated financial statements.
In August 2020, the FASB issued ASU No. 2020-06, Debt – Debt with Conversion and Other Options (Subtopic 470-20) and Derivatives and Hedging – Contracts in Entity’s Own Equity (Subtopic 815-40): Accounting for Convertible Instruments and Contracts in an Entity’s Own Equity, which simplifies the accounting for convertible instruments by eliminating the requirement to separate embedded conversion features from the host contract when the conversion features are not required to be accounted for as derivatives under Topic 815, Derivatives and Hedging, or that do not result in substantial premiums accounted for as paid-in capital. By removing the separation model, a convertible debt instrument will be reported as a single liability instrument with no separate accounting for embedded conversion features. This new standard also removes certain settlement conditions that are required for contracts to qualify for equity classification and simplifies the diluted earnings per share calculations by requiring that an entity use the if-converted method and that the effect of potential share settlement be included in diluted earnings per share calculations. The new standard will be effective for fiscal years beginning after December 15, 2023, for smaller reporting companies. As the Company currently does not have debt with conversion and other options, the Company does not believe the adoption will have a material impact on our consolidated financial statements.
Note 3. PREPAID EXPENSES AND OTHER CURRENT ASSETS
Prepaid expenses and other current assets at December 31, 2022 and 2021, are summarized below:
| December 31, | ||||||||
| 2022 | 2021 | |||||||
| Prepaid insurance | $ | $ | ||||||
| Legal and professional | ||||||||
| Other | ||||||||
| Total prepaid expenses and other current assets | $ | $ | ||||||
Note 4. PROPERTY AND EQUIPMENT, NET
Property and equipment at December 31, 2022 and 2021, are summarized below:
| December 31, | ||||||||
| 2022 | 2021 | |||||||
| Lab equipment | $ | $ | ||||||
| Computers and software | ||||||||
| Less: accumulated depreciation and amortization | ( |
) | ( |
) | ||||
| Total property and equipment, net | $ | $ | ||||||
Depreciation
and amortization expense was $
Note 5. ACCRUED EXPENSES
Accrued expenses at December 31, 2022 and 2021, are summarized below:
| December 31, | ||||||||
| 2022 | 2021 | |||||||
| Compensation | $ | $ | ||||||
| Legal and professional | ||||||||
| Clinical | ||||||||
| Other | ||||||||
| Total accrued expenses | $ | $ | ||||||
| F-20 |
bioAffinity Technologies, Inc.
Notes to Consolidated Financial Statements
For the Years Ended December 31, 2022 and 2021
Note 6. LOAN PAYABLE
The Coronavirus Aid, Relief, and Economic Security Act (the “CARES Act”) provided stimulus measures, including the Paycheck Protection Program (“PPP”), to provide certain small businesses with liquidity to support their operations during the COVID-19 pandemic.
In
April 2020, the Company received an initial $
In
March 2021, the Company received a second PPP Loan for $
In
September 2022, the Company obtained short-term financing of approximately $
Note 7. FAIR VALUE MEASUREMENTS
The Company analyzes all financial instruments with features of both liabilities and equity under the Financial Accounting Standard Board’s (“FASB”) accounting standard for such instruments. Under this standard, financial assets and liabilities are classified in their entirety based on the lowest level of input that is significant to the fair value measurement.
The estimated fair value of certain financial instruments, including cash and cash equivalents, accounts receivable, prepaid and other expenses, accounts payable, and accrued expenses are carried at historical cost basis, which approximates their fair values because of the short-term nature of these instruments. There are no assets and liabilities that are measured at fair value at December 31, 2022. The table below summarizes the Company’s assets and liabilities that are measured at fair value at December 31, 2021:
| Fair value measured at December 31, 2021 | ||||||||||||||||
|
Total at December 31, 2021 |
Quoted Prices in active markets |
Significant other observable inputs |
Significant unobservable inputs |
|||||||||||||
| Convertible notes payable | $ | $ | ||||||||||||||
A description of the valuation techniques and the values used for significant unobservable inputs to derive fair value measurements for those assets and liabilities measured at fair value at December 31, 2021:
| Fair value | Valuation technique |
Unobservable Input |
Range (weighted average) |
|||||||||
| Convertible notes payable at 12/31/21 | $ |
Risky Put + Stock Payoff |
Probability weighting assigned to automatic and optional conversion scenarios | %/ | % | |||||||
| Applied discount rate | % | |||||||||||
| Common share class volatility | % | |||||||||||
| Preferred stock class volatility | % | |||||||||||
| Negotiation discount | % | |||||||||||
| F-21 |
bioAffinity Technologies, Inc.
Notes to Consolidated Financial Statements
For the Years Ended December 31, 2022 and 2021
The
Company transferred $
| Fair value of convertible notes payable at December 31, 2020 | $ | |||
| Convertible notes payable issued | ||||
| Debt discount for warrants issued | ( |
) | ||
| Accretion of debt issuance costs | ||||
| Change in fair value of convertible notes payable | ( |
) | ||
| Fair value of convertible notes payable at December 31, 2021 | $ | |||
| Additional convertible notes payable issued | ||||
| Repayment of convertible notes payable | ( |
) | ||
| Debt discount for warrants issued | ( |
) | ||
| Accretion of debt issuance costs | ||||
| Change in fair value of convertible notes payable | ||||
| Transfer from level 3 to level 2 | ( |
) | ||
| Conversion of convertible notes payable into common stock | ( |
) | ||
| Fair value of convertible notes payable at December 31, 2022 | $ |
Note 8. CONVERTIBLE NOTES PAYABLE
In
September 2022, in connection with the closing of the IPO, the Company converted approximately $
From
August 2018 through July 2020, the Company issued a total of $
From
October 2020 through June 2021, the Company issued a total of $
In
the second and third quarters of 2021, the Company issued a total of approximately $
| F-22 |
bioAffinity Technologies, Inc.
Notes to Consolidated Financial Statements
For the Years Ended December 31, 2022 and 2021
Bridge Notes
In
the fourth quarter of 2021 and until our IPO in the third quarter of 2022, the Company issued a total of $
Additionally,
each noteholder received a warrant to purchase one share of Common Stock based on the investor’s bridge note principal balance
investment. The warrants have a term at an exercise price equal to $
The Company elected to account for the convertible notes payable at fair value with any changes in fair value being recognized through the consolidated statements of operations until the convertible notes are settled. The fair value of the convertible notes was determined with the assistance of a third-party specialist, considering the value of the notes payable that would be received by converting into common stock in each scenario, plus a put option. In coordination with the Company’s IPO, the notes were converted to Common Stock. Convertible notes payable consisted of the following:
| December 31, | ||||
| 2021 | ||||
| Secured convertible notes payable | $ | |||
| Unsecured convertible notes payable | ||||
| Principal amount of convertible notes payable | ||||
| Debt issuance costs | ( |
) | ||
| Fair value adjustments on convertible notes payable | ||||
| Total convertible notes payable | $ | |||
The Company elected to account for the convertible notes payable at fair value with any changes in fair value being recognized through the consolidated statements of operations until the convertible notes are settled. The fair value of the convertible notes was determined with the assistance of a third-party specialist, considering the value of the notes payable that would be received by converting into Common Stock in each scenario, plus a put option.
Note 9. COMMITMENTS AND CONTINGENCIES
Operating Leases
The
Company leases its corporate offices under a month-to-month agreement and lab space under an operating lease that is renewable annually
and expires in February 2024. Rent expense for office and lab space amounted to approximately $
Legal Matters
From time to time, the Company is involved in various disputes and litigation matters that arise in the ordinary course of business. To date, the Company had no material pending legal proceedings.
Note 10. CONVERTIBLE PREFERRED STOCK AND STOCKHOLDERS’ EQUITY (DEFICIT)
In
June 2022, the Company completed a
| F-23 |
bioAffinity Technologies, Inc.
Notes to Consolidated Financial Statements
For the Years Ended December 31, 2022 and 2021
Convertible Preferred Stock
The
Company has authorized a total of shares of $ per share par value preferred stock. In July 2017, the Company completed
a private placement of securities in which million shares of Series A preferred stock were sold, resulting in net proceeds of $
In
accordance with the Certificate of Designation of the Series A preferred stock, all of the shares of Series A preferred stock that were
issued and outstanding at the time of the IPO were automatically converted into fully paid and nonassessable shares of Common
Stock at a 1-for-7 conversion rate (as adjusted for the
The Company classifies convertible preferred stock outside of stockholders’ deficit because the shares contain deemed liquidation rights that are a contingent redemption feature not solely within the control of the Company. The holders of the Series A preferred stock had various rights, preferences, and privileges as follows:
Voting Rights
Dividends
Optional Conversion Rights
Each share of Series A preferred stock was convertible, at the option of the holder, at any time after the date of issuance of such share into such number of fully paid and nonassessable shares of Common Stock as is determined by dividing the Series A original issuance price by the conversion price in effect at the time of conversion. As of December 31, 2021, each of the shares of Series A preferred stock was convertible into one share of Common Stock. The respective applicable conversion prices for the Series A preferred stock were subject to adjustment upon any future stock split, stock dividend, combination, reclassification, or similar event affecting the convertible preferred stock or any series thereof.
Mandatory Conversion Rights
Each
share of Series A preferred stock automatically converted into the number of shares of Common Stock determined in accordance with the
conversion rate upon the earlier of: (a) the closing of a public offering of Common Stock at a price of at least $
Liquidation Preference
In the event of any liquidation, dissolution, or winding up of the Company, either voluntary or involuntary, the holders of Series A preferred stock were entitled to receive an amount equal to $ per share (subsequent to the reverse-stock-split calculation) plus an additional amount equal to any dividends declared or accrued but unpaid on each share. If, upon such liquidation event, the assets and funds distributed are insufficient to permit the payment to each holder of the Series A preferred stock of the full preferential amount, the entire assets and funds legally available for distribution to the holders of Series A preferred stock would have been distributed ratably among the holders of the Series A preferred stock based on the number of shares held. Deemed liquidation events include the sale of the Company or grant of an unlimited exclusive license to the Company’s technology or intellectual property rights.
| F-24 |
bioAffinity Technologies, Inc.
Notes to Consolidated Financial Statements
For the Years Ended December 31, 2022 and 2021
Common Stock
The Company has authorized a total of shares of $ per share par value Common Stock. Holders of Common Stock are entitled to cast one vote for each share held of record on all matters presented to the stockholders and have no cumulative voting rights. As of December 31, 2022, the Company has issued shares of Common Stock.
In November 2021, the Company received shareholder approval to increase the number of authorized shares from to a total of shares of $ per share par value Common Stock.
The Company grants options under its 2014 Equity Incentive Plan (the “Plan”). The Plan is authorized to grant Incentive Stock Options, Non-statutory Stock Options, or Restricted Stock for up to million shares of Common Stock, or twenty percent (20%) of the total issued and outstanding Common Stock, whichever is greater. The Company has reserved million shares to be under the plan. Options may be granted to employees, the Company’s board of directors, and external consultants who provide service to the Company. The options have vesting schedules with terms of one to four years and become fully exercisable based on specific terms imposed at the date of grant. The requisite service period for employees or consultants begins on the grant date and ends when the employee or consultant ceases to be employed or providing service, unless a longer period is provided in the option agreement. The requisite service period for directors begins on the grant date and ends on the option term provided in the option agreement. Options are exercisable for a period of up to ten (10) years from grant date. The Plan will terminate according to the respective terms of the Plan in September 2026.
| 2022 | 2021 | |||||||
| Research and development | $ | $ | ||||||
| Selling, general and administrative | ||||||||
| Total stock-based compensation expense | $ | $ | ||||||
| Number of options |
Weighted- average exercise price |
Weighted- average remaining contractual term (in years) |
Aggregate intrinsic value |
|||||||||||||
| Outstanding at December 31, 2020 | $ | |||||||||||||||
| Granted | ||||||||||||||||
| Exercised | ||||||||||||||||
| Forfeited | ( |
) | ||||||||||||||
| Outstanding at December 31, 2021 | $ | |||||||||||||||
| Granted | ||||||||||||||||
| Exercised | ( |
) | ||||||||||||||
| Forfeited | ( |
) | ||||||||||||||
| Outstanding at December 31, 2022 | $ | $ | ||||||||||||||
| Vested and exercisable at December 31, 2022 | $ | $ | ||||||||||||||
As of December 31, 2022, there was unrecognized compensation cost related to non-vested stock options.
During the year ended December 31, 2021, the Company issued options to purchase shares of Common Stock to employees and non-employees. The per share weighted-average fair value of the options granted during 2021 was estimated at $ on the date of grant. During the year ended December 31, 2021, options were exercised.
During the year ended December 31, 2021, the Company issued restricted stock units (RSUs) for shares of Common Stock to employees. The shares vest in equal monthly installments over terms of between one to three years, subject to the employee providing continuous service through the vesting date. The approximately unissued shares vest over a weighted-average period of years.
During
the year ended December 31, 2022, the Company issued options to purchase shares of Common Stock to employees. The per share weighted-average
fair value of the options granted during 2022 was estimated at $ on the date of grant. During the year ended December 31, 2022,
options were exercised into an equivalent number of common shares. The company received proceeds of approximately $
| F-25 |
bioAffinity Technologies, Inc.
Notes to Consolidated Financial Statements
For the Years Ended December 31, 2022 and 2021
| 2022 | 2021 | |||||||
| Fair value of Common Stock | $ | $ | ||||||
| Volatility | % | % | ||||||
| Expected term (years) | ||||||||
| Risk-free interest rate | % | % | ||||||
| Dividend yield | % | % | ||||||
Black-Scholes requires the use of subjective assumptions which determine the fair value of stock-based awards. These assumptions include:
Fair value of Common Stock—The fair value of stock option and restricted share grants are determined based on the closing price of our stock on the date of grant.
Expected term—The expected term represents the period that stock-based awards are expected to be outstanding. The expected term for option grants is determined using the simplified method. The simplified method deems the term to be the average of the time-to-vesting and the contractual life of the stock-based awards.
Expected volatility— Since the Company does not have sufficient trading history for its Common Stock, the expected volatility is estimated based on the average volatility for comparable publicly traded biotechnology companies over a period equal to the expected term of the stock-based awards. The comparable companies were chosen based on their similar size, stage in the life cycle or area of specialty. The Company will continue to apply this process until a sufficient amount of historical information regarding the volatility of its own stock price becomes available.
Risk-free interest rate—The risk-free interest rate is based on the U.S. Treasury zero coupon issues in effect at the time of grant for periods corresponding with the expected term of a stock-based award.
Expected dividend—The Company has never paid dividends on its Common Stock and has no plans to pay dividends on its Common Stock. Therefore, the Company used an expected dividend yield of zero.
Note 12. WARRANTS
We account for Common Stock warrants as either equity instruments or derivative liabilities depending on the specific terms of the warrant agreement. Warrants are accounted for as derivative liabilities if the warrants allow for cash settlement or provide for modification of the warrant exercise price in the event subsequent sales of Common Stock by the Company are at a lower price per share than the then-current warrant exercise price. We classify derivative warrant liabilities on the consolidated balance sheet at fair value, and changes in fair value during the periods presented in the consolidated statement of operations, which is revalued at each consolidated balance sheet date subsequent to the initial issuance of the stock warrant.
In
September 2022, in connection with our IPO, we issued a total of
| F-26 |
bioAffinity Technologies, Inc.
Notes to Consolidated Financial Statements
For the Years Ended December 31, 2022 and 2021
In
2022,
In
2022, the Company issued an additional
From
October 2021 through August 2022, the Company issued approximately $
In
2021, the Company issued an aggregate of
In
connection with the issuance of the Bridge Notes, the Company amended the 2018 and 2020 Notes whereby upon completion of an IPO, all
outstanding principal and interest will convert into shares of the Company’s Common Stock and at $
The following table summarizes the calculated aggregate fair values for the warrant derivative liability using the Black-Scholes method based on the following assumptions at December 31, 2022:
| Exercise price per share of warrant | $ | |||
| Fair market closing price per share of Common Stock | $ | |||
| Volatility | % | |||
| Expected term (years) | ||||
| Risk-free interest rate | % | |||
| Dividend yield | % |
In
March 2017, the Company issued an aggregate of
Note 13. INCOME TAXES
Deferred tax assets and valuation allowance
The
Company had, subject to limitation, approximately $
| December 31, | ||||||||
| 2022 | 2021 | |||||||
| Deferred tax assets: | ||||||||
| Net operating loss carryover | $ | $ | ||||||
| Stock compensation | ||||||||
| Capitalized R&E costs | ||||||||
| Depreciation and amortization | ( |
) | ||||||
| Other | ||||||||
| Tax credits | ||||||||
| Total deferred tax assets | ||||||||
| Less: valuation allowance | ( |
) | ( |
) | ||||
| Net deferred tax assets | $ | $ | ||||||
| F-27 |
bioAffinity Technologies, Inc.
Notes to Consolidated Financial Statements
For the Years Ended December 31, 2022 and 2021
The reconciliation of the statutory federal income tax rate to the Company’s effective tax rate for the years ended
December 31, 2022 and 2021, was as follows:
| Year Ended December 31, | ||||||||
| 2022 | 2021 | |||||||
| Tax at federal statutory rate | ( |
)% | ( |
)% | ||||
| Permanent differences | ||||||||
| Research and development credits | ( |
) | ||||||
| Change in valuation allowance | ||||||||
| Effective income tax rate | % | % | ||||||
Unrecognized tax benefits
As
of December 31, 2022 and 2021, the Company has unrecognized tax benefits related to tax credits of $
| December 31, | ||||||||
| 2022 | 2021 | |||||||
| Beginning balance | $ | $ | ||||||
| Deductions based on tax positions related to the prior year | ( |
) | ||||||
| Additions based on tax positions related to the current year | ||||||||
| Ending balance | $ | $ | ||||||
The Company is not under audit with any taxing jurisdiction at this time. The Company’s tax returns for the previous three years remain open for audit by the respective tax jurisdictions.
Note 14. RELATED PARTY TRANSACTIONS
From
August 2018 through July 2020, the Company has issued a total of $
All
of these notes bore interest at
In
August 2022, Maria Zannes, the founder, President, Chief Executive Officer, and a director of the Company, purchased a Bridge Note in
the principal amount of $
In
August 2022, Steven Girgenti, the Executive Chairman and a director of the Company, purchased a Bridge Note in the principal amount of
$
Note 15. SUBSEQUENT EVENTS
The Company evaluated all events or transactions that occurred after December 31, 2022, up through the date the consolidated financial statements were issued. During this period, the Company did not have any material subsequent events required to be disclosed as of and for the period ended December 31, 2022.
| F-28 |
Independent Auditor’s Report
To the Board of Directors and Stockholders of
Village Oaks Pathology Services, P.A.:
Opinion
We have audited the financial statements of Village Oaks Pathology Services, P.A. (the “Company”), which comprise the balance sheets as of December 31, 2022 and 2021, and the related statements of operations, changes in stockholders’ equity and cash flows for the years then ended, and the related notes to the financial statements.
In our opinion, the accompanying financial statements present fairly, in all material respects, the financial position of Village Oaks Pathology Services, P.A. as of December 31, 2022 and 2021, and the results of its operations and its cash flows for the years then ended in accordance with accounting principles generally accepted in the United States of America.
Basis of Opinion
We conducted our audits in accordance with auditing standards generally accepted in the United States of America (“GAAS”). Our responsibilities under those standards are further described in the Auditor’s Responsibilities for the Audit of the Financial Statements section of our report. We are required to be independent of the Company and to meet our other ethical responsibilities, in accordance with the relevant ethical requirements relating to our audits. We believe that the audit evidence we have obtained is sufficient and appropriate to provide a basis for our audit opinion.
Substantial Doubt About the Entity’s Ability to Continue as a Going Concern
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 2 to the financial statements, the Company has recurring and expected losses from operations, negative operating cash flows, and is projecting its cash outflows to exceed its cash on hand over the next twelve months. These factors raise substantial doubt regarding the Company’s ability to continue as a going concern. Management’s evaluation of the events and conditions and management’s plans regarding these matters are also described in Note 2. The financial statements do not include any adjustments that might result from the outcome of this uncertainty. Our opinion is not modified with respect to this matter.
Responsibilities of Management for the Financial Statements
Management is responsible for the preparation and fair presentation of the financial statements in accordance with accounting principles generally accepted in the United States of America, and for the design, implementation, and maintenance of internal control relevant to the preparation and fair presentation of financial statements that are free from material misstatement, whether due to fraud or error.
In preparing the financial statements, management is required to evaluate whether there are conditions or events, considered in the aggregate, that raise substantial doubt about the Company’s ability to continue as a going concern for one year after the date that the financial statements are available to be issued.
Auditor’s Responsibilities for the Audit of the Financial Statements
Our objectives are to obtain reasonable assurance about whether the financial statements as a whole are free from material misstatement, whether due to fraud or error, and to issue an auditor’s report that includes our opinion. Reasonable assurance is a high level of assurance but is not absolute assurance and therefore is not a guarantee that an audit conducted in accordance with GAAS will always detect a material misstatement when it exists. The risk of not detecting a material misstatement resulting from fraud is higher than for one resulting from error, as fraud may involve collusion, forgery, intentional omissions, misrepresentations, or the override of internal control. Misstatements are considered material if there is a substantial likelihood that, individually or in the aggregate, they would influence the judgment made by a reasonable user based on the financial statements.
In performing an audit in accordance with GAAS, we:
| ● | Exercise professional judgment and maintain professional skepticism throughout the audit. | |
| ● | Identify and assess the risks of material misstatement of the financial statements, whether due to fraud or error, and design and perform audit procedures responsive to those risks. Such procedures include examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. | |
| ● | Obtain an understanding of internal control relevant to the audit in order to design audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control. Accordingly, no such opinion is expressed. | |
| ● | Evaluate the appropriateness of accounting policies used and the reasonableness of significant accounting estimates made by management, as well as evaluate the overall presentation of the financial statements. | |
| ● | Conclude whether, in our judgment, there are conditions or events, considered in the aggregate, that raise substantial doubt about the Company’s ability to continue as a going concern for a reasonable period of time. |
We are required to communicate with those charged with governance regarding, among other matters, the planned scope and timing of the audit, significant audit findings, and certain internal control–related matters that we identified during the audit.
Emphasis of Matter
As described in Note 2 to the financial statements, the Company adopted Accounting Standards Codification (“ASC”) Topic 842, Leases, as of January 1, 2022. The prior year financial statements have not been adjusted and continue to be reported in accordance with the Company’s historic accounting under ASC Topic 840. Our opinion is not modified with respect to this matter.
/s/ WithumSmith+Brown, PC
September 19, 2023
| F-29 |
VILLAGE OAKS PATHOLOGY SERVICES, P.A.
D/B/A PRECISION PATHOLOGY SERVICES
BALANCE SHEETS
| As of December 31, | ||||||||
| 2022 | 2021 | |||||||
| ASSETS | ||||||||
| Current Assets | ||||||||
| Cash | $ | 357,470 | $ | 1,207,341 | ||||
| Certificates of deposit | 100,823 | 100,722 | ||||||
| Investments | 259,392 | 300,051 | ||||||
| Patient fees receivable | 858,950 | 696,759 | ||||||
| Other receivables | 381,204 | 515,464 | ||||||
| Prepaid expenses | 31,123 | 3,374 | ||||||
| Total Current Assets | 1,988,962 | 2,823,711 | ||||||
| Non-Current Assets | ||||||||
| Property and equipment, net | 328,861 | 283,777 | ||||||
| Operating lease right-of-use asset, net | 494,900 | — | ||||||
| Finance/capital lease right-of-use asset, net | 1,554,889 | 1,491,014 | ||||||
| Deposits | 8,000 | 8,000 | ||||||
| Total Non-Current Assets | 2,386,650 | 1,782,791 | ||||||
| TOTAL ASSETS | $ | 4,375,612 | $ | 4,606,502 | ||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY | ||||||||
| Current Liabilities | ||||||||
| Accounts payable | $ | 83,386 | $ | 80,136 | ||||
| Accrued expenses | 351,276 | 237,216 | ||||||
| Notes payable, current portion | 40,407 | 33,647 | ||||||
| Operating lease liability, current portion | 96,654 | — | ||||||
| Finance/capital lease liability, current portion | 413,729 | 367,680 | ||||||
| Total Current Liabilities | 985,452 | 718,679 | ||||||
| Non-Current Liabilities | ||||||||
| Notes payable, net of current portion | 95,879 | 100,100 | ||||||
| PPP loan payable | — | 503,950 | ||||||
| Deferred rent | — | 14,078 | ||||||
| Operating lease liability, net of current portion | 403,177 | — | ||||||
| Finance/capital lease liability, net of current portion | 1,218,535 | 1,123,334 | ||||||
| Total Non-Current Liabilities | 1,717,591 | 1,741,462 | ||||||
| TOTAL LIABILITIES | 2,703,043 | 2,460,141 | ||||||
| Commitments and contingencies (see Note 11) | ||||||||
| Stockholders’ Equity | ||||||||
| Common stock, authorized 1,000, $0.01 par value; 500 shares issued and outstanding as of December 31, 2022 and 2021 | 5 | 5 | ||||||
| Retained earnings | 1,672,564 | 2,146,356 | ||||||
| Total Stockholders’ Equity | 1,672,569 | 2,146,361 | ||||||
| TOTAL LIABILITIES AND STOCKHOLDERS’ EQUITY | $ | 4,375,612 | $ | 4,606,502 | ||||
See accompanying notes to the financial statements.
| F-30 |
VILLAGE OAKS PATHOLOGY SERVICES, P.A.
D/B/A PRECISION PATHOLOGY SERVICES
STATEMENTS OF OPERATIONS
| Years Ended December 31, | ||||||||
| 2022 | 2021 | |||||||
| Net Revenue | $ | 6,858,212 | $ | 6,196,631 | ||||
| Operating Expenses | ||||||||
| Selling, general, and administrative | 7,184,802 | 5,757,341 | ||||||
| Depreciation and amortization | 544,217 | 536,481 | ||||||
| Total Operating Expenses | 7,729,019 | 6,293,822 | ||||||
| Loss from Operations | (870,807 | ) | (97,191 | ) | ||||
| Other Income (Expense) | ||||||||
| PPP loan forgiveness | 503,950 | 503,900 | ||||||
| Other income, net | 9,192 | 12,467 | ||||||
| Interest expense | (63,308 | ) | (12,730 | ) | ||||
| Investment income | 8,775 | 4,396 | ||||||
| Unrealized loss on investments | (49,434 | ) | (4,345 | ) | ||||
| Total Other Income | 409,175 | 503,688 | ||||||
| Net income (loss) | $ | (461,632 | ) | $ | 406,497 | |||
See accompanying notes to the financial statements.
| F-31 |
VILLAGE OAKS PATHOLOGY SERVICES, P.A.
D/B/A PRECISION PATHOLOGY SERVICES
STATEMENTS OF STOCKHOLDERS’ EQUITY
| Retained Earnings | Common Stock | Total Stockholders’ Equity |
||||||||||
| Balance as of December 31, 2020 | $ | 1,751,859 | $ | 5 | $ | 1,751,864 | ||||||
| Distributions | (12,000 | ) | — | (12,000 | ) | |||||||
| Net income | 406,497 | — | 406,497 | |||||||||
| Balance as of December 31, 2021 | $ | 2,146,356 | $ | 5 | $ | 2,146,361 | ||||||
| Distributions | (12,160 | ) | — | (12,160 | ) | |||||||
| Net loss | (461,632 | ) | — | (461,632 | ) | |||||||
| Balance as of December 31, 2022 | $ | 1,672,564 | $ | 5 | $ | 1,672,569 | ||||||
See accompanying notes to the financial statements.
| F-32 |
VILLAGE OAKS PATHOLOGY SERVICES, P.A.
D/B/A PRECISION PATHOLOGY SERVICES
STATEMENTS OF CASH FLOWS
| Years Ended December 31, | ||||||||
| 2022 | 2021 | |||||||
| Cash flows from operating activities: | ||||||||
| Net income (loss) | $ | (461,632 | ) | $ | 406,497 | |||
| Adjustments to reconcile net income (loss) to net cash provided by (used in) operating activities: | ||||||||
| Forgiveness of PPP loan payable | (503,950 | ) | (503,900 | ) | ||||
| Depreciation and amortization | 544,217 | 536,481 | ||||||
| Loss on disposal of fixed assets | — | 16,784 | ||||||
| Investment income | (8,775 |
) | (4,396 |
) | ||||
| Unrealized loss on investments | 49,434 | 4,345 | ||||||
| Changes in operating assets and liabilities: | ||||||||
| Patient fees receivable | (162,191 | ) | (50,077 | ) | ||||
| Other receivables | 134,260 | (8,362 | ) | |||||
| Prepaid expenses | (27,749 | ) | (2,481 | ) | ||||
| Accounts payable | 3,250 | 59,455 | ||||||
| Accrued expenses | 114,060 | 51,054 | ||||||
| Operating lease liability | (9,147 | ) | — | |||||
| Deferred rent | — | (326 |
) | |||||
| Net cash provided by (used in) operating activities | (328,223 | ) | 505,074 | |||||
| Cash flows from investing activities: | ||||||||
| (Purchase) sale of certificates of deposit | (101 | ) | 302,835 | |||||
| Purchase of investments | — | (300,000 | ) | |||||
| Purchase of property and equipment | (144,246 | ) | (192,921 | ) | ||||
| Net cash used in investing activities | (144,347 | ) | (190,086 | ) | ||||
| Cash flows from financing activities: | ||||||||
| Borrowings of notes payable | 39,953 | 93,414 | ||||||
| Repayments of notes payable | (37,414 | ) | (36,094 | ) | ||||
| Proceeds from PPP loan payable | — | 503,950 | ||||||
| Net payments from financing lease | (367,680 | ) | (505,651 | ) | ||||
| Distributions | (12,160 | ) | (12,000 | ) | ||||
| Net cash provided by (used in) financing activities | (377,301 | ) | 43,619 | |||||
| Net change in cash | (849,871 | ) | 358,607 | |||||
| Cash, beginning of year | 1,207,341 | 848,734 | ||||||
| Cash, end of year | $ | 357,470 | $ | 1,207,341 | ||||
| Supplemental disclosure of cash flow information: | ||||||||
| Cash paid for interest | $ | 63,308 | $ | 12,730 | ||||
| Supplemental disclosure of noncash investing and financing activities: | ||||||||
| Equipment purchase included in accounts payable | $ | — | $ | 12,995 | ||||
| Operating lease liabilities arising from obtaining right-of-use assets | 590,474 | — | ||||||
| Finance lease liabilities arising from right-of-use asset modification | 508,930 | — | ||||||
See accompanying notes to the financial statements.
| F-33 |
VILLAGE OAKS PATHOLOGY SERVICES, P.A.
D/B/A PRECISION PATHOLOGY SERVICES
NOTES TO FINANCIAL STATEMENTS
1. Nature of Operations
Village Oaks Pathology Services, P.A., doing business as Precision Pathology Services (the “Company” or “Precision Pathology”) is a privately held company organized in 1987 under the laws of the state of Texas. Precision Pathology provides anatomic and clinical pathology services for patients and their physicians. The Company is known for their exceptionally responsive and helpful service to the physicians and patients they serve.
Income Taxes
The Company, with stockholders’ consent, has elected to be taxed as an “S Corporation” under the provisions of the Internal Revenue Code and comparable state income tax law. As an S Corporation, the Company is generally not subject to corporate income taxes and the Company’s net income or loss is reported on the individual tax return of the stockholders of the Company. Therefore, no provision or liability for income taxes is reflected in the financial statements. The Company has not been audited by the Internal Revenue Service, and accordingly the business tax returns since 2020 are open to examination. Management has evaluated its tax positions and has concluded that the Company had taken no uncertain tax positions that could require adjustment or disclosure in the financial statements to comply with provisions set forth in Accounting Standards Codification (“ASC”) Topic 740, Income Taxes.
2. Summary of Significant Accounting Policies
Basis of Presentation
The accompanying financial statements are prepared in accordance with generally accepted accounting principles in the United States of America (“U.S. GAAP”).
Use of Estimates
The preparation of financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. The Company evaluates estimates and assumptions on a regular basis. The Company bases its estimates and assumptions on current facts, historical experience and various other factors that it believes to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities and the accrual of costs and expenses that are not readily apparent from other sources. The Company’s accounting policies that involve significant judgment and estimates include revenue recognition including contractual adjustments and discounts, patient fee receivables and the related allowance for contractual discounts and allowance for doubtful accounts, valuation of lease liabilities and related right-of-use assets, and estimates of useful lives for depreciation. The actual results experienced by the Company may differ materially and adversely from the Company’s estimates. To the extent there are material differences between the estimates and the actual results, future results of operations will be affected.
Liquidity and Capital Resources
In accordance with Accounting Standards Update (“ASU”) 2014-15, Presentation of Financial Statements – Going Concern (Subtopic 205-40), the Company has evaluated whether there are conditions and events that raise substantial doubt about the Company’s ability to continue as a going concern for at least one year after the date the financial statements are issued. As required by this standard, management’s evaluation shall initially not take into consideration the potential mitigating effects of management’s plans that have not been fully implemented as of the date the financial statements are issued.
The Company’s assessment included the preparation of a detailed cash forecast that included all projected cash inflows and outflows. Although the Company continues to focus on growing its revenues, the Company’s ongoing operating expenditures will exceed the revenue it expects to receive for the foreseeable future. Additionally, the Company has a history of operating losses and negative operating cash flows and expects these trends to continue. Our future plans may include cash flows generated from our revenues, issuance of debt, or sales of our equity securities.
The Company’s loss from operations before depreciation and amortization was ($326,590) for the fiscal year ended December 31, 2022. The Company’s cash, certificates of deposit and investments and net working capital at December 31, 2022, were $717,685 and $1,003,510, respectively. Based on the Company’s current expected level of operating expenditures and continued revenue projections, the Company does not believe its cash on hand is sufficient to fund the Company’s ongoing operations for a period of a least twelve (12) months; therefore, the Company has concluded there is substantial doubt about the Company’s ability to continue as a going concern for one year from the issuance of these financial statements. Despite a history of successfully implementing similar plans to alleviate adverse financial conditions, these sources of working capital are not currently assured, and consequently do not sufficiently mitigate the risks and uncertainties disclosed above. These financial statements do not include any adjustments to reflect the possible future effects on the recoverability and classification of assets or the amounts of liabilities that may result from uncertainty related to the Company’s ability to continue as a going concern.
| F-34 |
VILLAGE OAKS PATHOLOGY SERVICES, P.A.
D/B/A PRECISION PATHOLOGY SERVICES
NOTES TO FINANCIAL STATEMENTS
Cash
The Company’s cash is held with one financial institution, and the account balances may exceed the Federal Deposit Insurance Corporation (“FDIC”) insurance limit at times. Accounts are insured by the FDIC up to $250,000. As of December 31, 2022 and 2021, the Company had uninsured cash deposits of $107,470 and $957,341, respectively. The Company has not experienced any losses in such accounts to date. Any loss incurred or a lack of access to such funds could have a significant adverse impact on the Company’s financial condition, results of operations, and cash flows. All highly liquid investments with maturities of three months or less at the date of purchase are classified as cash equivalents.
Investments held in MML Investors Services Account
At December 31, 2022 and 2021, the assets held in the MML Investors Services Account (“MML Account”) were held in money market funds, which are invested in fixed income and equity securities with balances of $259,392 and $300,051, respectively. Trading securities are presented on the balance sheet at fair value at the end of each reporting period. Gains and losses resulting from the change in fair value of these securities is included in unrealized loss on investments in the accompanying statements of operations. Dividend income and short-term and long-term capital gains on these securities is included in investment income in the accompanying statements of operations.
Certificates of Deposit
The Company invests its excess cash in bank certificates of deposit (“CDs”) which are fully insured by the FDIC with terms of not more than six months. As of December 31, 2022 and 2021, the Company had certificates of deposit with balances of $100,823 and $100,722, respectively.
Patient Fees Receivable
Patient accounts receivable represents amounts due from patient services billed to commercial insurance companies, governmental payors, and patients. Receivables are recorded at the amount the Company expects to collect. The Company estimates variable consideration for patient service fees using an expected value method. Accordingly, the Company has developed ratios for portfolios of payors based on the nature of the payor (e.g., commercial insurer, government program, uninsured patients), which impacts the average time to collect the consideration to which the Company expects to be entitled and the amount of such consideration. The Company has developed payment-to-charge ratio for each portfolio of payor based on historical payment experience and applied those ratios to gross charges for each year presented in order to arrive at the net patient fees receivable.
Other Receivables
Other receivables represent amounts billed for pathologist interpretations and medical director fees, which include the Company’s pathologists providing directorship for certain hospital facilities. Other receivables are recorded at the amount the Company expects to collect. Management determines uncollectible amounts based on historical collection experience. As of December 31, 2022 and 2021, management determined no allowance was necessary related to these receivables.
Property and Equipment, net
In accordance with Accounting Standards Codification (“ASC”) 360-10, Accounting for the Impairment of Long- Lived Assets, the Company periodically reviews the carrying value of its long-lived assets, such as property and equipment, to test whether current events or circumstances indicate that such carrying value may not be recoverable. When evaluating assets for potential impairment, the Company compares the carrying value of the asset to its estimated undiscounted future cash flows. If an asset’s carrying value exceeds such estimated cash flows (undiscounted and with interest charges), the Company records an impairment charge for the difference. The Company did not record impairment for the years ended December 31, 2022 and 2021.
Property and equipment are carried at cost, net of accumulated depreciation. Depreciation is computed using the straight-line method over the estimated useful life of the asset. Amortization of leasehold improvements is computed using the shorter of the lease term or estimated useful life of the asset. Additions and improvements are capitalized, while repairs and maintenance are expensed as incurred. Useful lives of each asset class are as follows:
| Asset Category | Useful Life | |
| Computer equipment | 5 years | |
| Computer software | 3 years | |
| Equipment | 5-7 years | |
| Furniture and fixtures | 5-7 years | |
| Vehicles | 5 years | |
| Leasehold improvements | Lesser of lease term or useful life |
| F-35 |
VILLAGE OAKS PATHOLOGY SERVICES, P.A.
D/B/A PRECISION PATHOLOGY SERVICES
NOTES TO FINANCIAL STATEMENTS
Fair Value of Financial Instruments
The Company applies ASC Topic 820, Fair Value Measurement (“ASC 820”), which establishes a framework for measuring fair value and clarifies the definition of fair value within that framework. ASC 820 defines fair value as an exit price, which is the price that would be received for an asset or paid to transfer a liability in the Company’s principal or most advantageous market in an orderly transaction between market participants on the measurement date. The fair value hierarchy established in ASC 820 generally requires an entity to maximize the use of observable inputs and minimize the use of unobservable inputs when measuring fair value. Observable inputs reflect the assumptions that market participants would use in pricing the asset or liability and are developed based on market data obtained from sources independent of the reporting entity. Unobservable inputs reflect the entity’s own assumptions based on market data and the entity’s judgments about the assumptions that market participants would use in pricing the asset or liability and are to be developed based on the best information available in the circumstances.
The carrying amounts reflected in the balance sheet for current assets and liabilities approximate fair value due to their short-term nature.
Level 1 — Assets and liabilities with unadjusted, quoted prices listed on active market exchanges. Inputs to the fair value measurement are observable inputs, such as quoted prices in active markets for identical assets or liabilities.
Level 2 — Inputs to the fair value measurement are determined using prices for recently traded assets and liabilities with similar underlying terms, as well as direct or indirect observable inputs, such as interest rates and yield curves that are observable at commonly quoted intervals.
Level 3 — Inputs to the fair value measurement are unobservable inputs, such as estimates, assumptions, and valuation techniques when little or no market data exists for the assets or liabilities.
See Note 4 for additional information on assets measured at fair value.
Revenue Recognition
The Company derives revenues from providing pathology testing services to patients and other customers. Revenue from services is recognized upon the transfer of control, which is generally achieved when testing is completed and the results are delivered to a patient, a patient’s physician, or institutional customers such as independent laboratories, hospitals, or contract research organizations (“CRO”). The Company’s revenues fall into three separate streams: (a) patient service fees, (b) histology service fees, and (c) medical director fees.
On January 1, 2021, the Company adopted ASC 606, Revenue from Contracts with Customers (“ASC 606”), using the modified retrospective method with respect to all non-completed contracts. ASC 606 outlines a single comprehensive model for entities to use in accounting for revenue arising from contracts with customers and supersedes nearly all existing revenue recognition guidance, including industry-specific guidance.
The new guidance is based on the principle that an entity should recognize revenue to depict the transfer of products or services to customers in an amount that reflects the consideration to which the entity expects to be entitled in exchange for those products or services. The adoption of ASC 606 did not have a material effect on the Company’s financial position, results of operations, or internal controls over financial reporting.
The Company determines revenue recognition by applying the following steps prescribed under ASC 606:
a. Identification of the contract, or contracts, with a customer;
b. Identification of the performance obligations in the contract;
c. Determination of the transaction price;
d. Allocation of the transaction price to the performance obligations in the contract; and
e. Recognition of revenue when, or as, we satisfy a performance obligation.
The Company collects patient service fees from patients and various third-party payors, mainly insurance companies and governmental payors. Patient service fees are earned from performing pathology lab services (procedures or tests), which may be requested by a patient directly or by a physician on a patient’s behalf. The Company also provides histology services to hospitals, CRO’s or independent laboratories. The Company’s services represent performance obligations transferred to the customer at the point in time when the test results are delivered, which is when the customer obtains the benefits of the service. Patient service fee revenue is variable given various factors that impact whether third-party payors ultimately pay the Company’s contractual billing rates. While third-party payor rates are known at inception of the contract, the payor has the ultimate discretion to adjudicate claims and decide on the final payment amount. There are various factors that allow third-party payors the right to deny all or part of a claim, which may not be known at inception of the contract. While the Company may appeal claim denials or adjustments, generally the Company offers some level of implicit price concession as part of these adjustments made by payors. Furthermore, patient service fees billed to uninsured patients is subject to variability for factors not known at inception. In contrast, the transaction price for histology services is generally fixed, as no third-party payors are involved, and therefore, the fees agreed upon upfront are the fees that the Company expects to collect for services performed.
The Company estimates variable consideration for patient service fees using an expected value method. Accordingly, the Company has developed ratios for portfolios of payors based on the nature of the payor (e.g., commercial insurer, government program, uninsured patients), which impacts the average time to collect the consideration to which the Company expects to be entitled and the amount of such consideration. The Company has developed payment-to-charge ratio for each portfolio of payor based on historical payment experience and applied those ratios to gross charges for each year presented. Variable consideration is constrained to the extent that it is deemed probable that a significant reversal in the amount of revenue recognized will not occur when the uncertainty is resolved, which is when an insurance claim is fully resolved.
| F-36 |
VILLAGE OAKS PATHOLOGY SERVICES, P.A.
D/B/A PRECISION PATHOLOGY SERVICES
NOTES TO FINANCIAL STATEMENTS
Advertising
Advertising costs are expensed as incurred. Advertising costs were $2,802 and $2,764 for the years ended December 31, 2022 and 2021, respectively, which are included in selling, general and administrative expense on the accompanying statements of operations.
Leases
The Company determines if an arrangement is a lease at inception and classifies its leases at commencement. Operating leases are presented as right-of-use (“ROU”) assets and the corresponding lease liabilities are included in operating lease liabilities, current and operating lease liabilities on the Company’s balance sheets. ROU assets represent the Company’s right to use an underlying asset, and lease liabilities represent the Company’s obligation for lease payments in exchange for the ability to use the asset for the duration of the lease term.
ROU assets and lease liabilities are recognized at the lease commencement date and determined using the present value of the future minimum lease payments over the lease term. The Company used a discount rate based on a benchmark approach as of January 1, 2022, the date of initial application of the new guidance, to derive an appropriate incremental borrowing rate to discount remaining lease payments. The Company benchmarked itself against other companies of similar credit ratings and comparable quality and derived imputed rates for lease term lengths ranging from approximately 1.9 to 5.6 years. The lease term may include options to extend when it is reasonably certain that the Company will exercise that option. In addition, the Company does not recognize short-term leases that have a term of twelve months or less as ROU assets or lease liabilities. The Company recognizes operating lease expense on a straight-line basis over the lease term.
The Company has lease agreements that contain both lease and non-lease components, which it has elected to account for as a single lease component when the payments are fixed. As such, variable lease payments, including those not dependent on an index or rate, such as real estate taxes, common area maintenance, and other costs that are subject to fluctuation from period to period are not included in lease measurement.
Recent Accounting Pronouncements
In February 2016, the FASB established Topic 842, Leases, by issuing ASU No. 2016-02 (“ASU 2016-02”), which requires lessees to recognize leases on balance sheet and disclose key information about leasing arrangements. The new standard establishes a right-of-use model that requires a lessee to recognize a ROU asset and lease liability on the balance sheet for all leases with a term longer than 12 months. Leases are classified as finance or operating, with classification affecting the pattern and classification of expense recognition in the income statement.
In June 2020, the FASB issued ASU No. 2020-05 (“ASU 2020-05”) which pushed back the effective date one year for private and not-for-profit entities that did not issue or serve as conduit bond obligors and had not yet adopted the standard. The new effective date was for fiscal year periods beginning after December 15, 2021.
The Company adopted ASU 2016-02 effective January 1, 2022, using a modified retrospective approach at the beginning of the year of adoption. In addition, the Company elected the transition package of three practical expedients permitted within the standard, which eliminates the requirements to reassess prior conclusions about lease identification, lease classification and initial direct costs. Further, the Company adopted a short-term lease exception policy, permitting the Company to not apply the recognition requirements of this standard to short-term leases (i.e., leases with terms of 12 months or less) and an accounting policy to account for lease and non-lease components as a single component for certain classes of assets. On January 1, 2022, the Company recorded lease liabilities and corresponding right-of-use assets based on the present value of the remaining minimum rental payments for leases existing upon adoption of the new lease standard and other adjustments to the opening balance of right-of-use assets, if any. The adoption of ASC 842 did not result in a material impact to the consolidated statements of operations or cash flows. See Note 7 for additional detail on the Company’s leasing arrangements.
In November 2018, the FASB issued ASU 2018-19, Codification Improvements to Topic 326, Financial Instruments—Credit Losses, which amends the guidance for accounting for assets that are potentially subject to credit risk. The amendments affect contract assets, loans, debt securities, trade receivables, net investments in leases, off-balance-sheet credit exposures, reinsurance receivables, and any other financial assets not excluded from the scope that have the contractual right to receive cash. ASU 2018-19 is effective for fiscal years beginning after December 15, 2022. The Company has not yet evaluated the accounting, transition and disclosure requirements of the ASU and cannot currently estimate the financial statement impact of adoption.
| F-37 |
VILLAGE OAKS PATHOLOGY SERVICES, P.A.
D/B/A PRECISION PATHOLOGY SERVICES
NOTES TO FINANCIAL STATEMENTS
3. Revenue, Net
The following is a summary of net revenue for the years ended December 31:
| 2022 | 2021 | |||||||
| Patient fees | $ | 5,378,629 | $ | 4,860,703 | ||||
| Histology service fees | 1,366,789 | 1,091,285 | ||||||
| Medical director fees | 103,119 | 98,887 | ||||||
| Other revenue | 9,675 | 145,756 | ||||||
| Revenue, net | $ | 6,858,212 | $ | 6,196,631 | ||||
4. Investments
The following table presents information about the Company’s financial assets and liabilities that are measured at fair value as of December 31, 2022 and 2021, and indicates the fair value hierarchy of the valuation inputs the Company utilized to determine such fair value:
| Fair Value | Level 1 | Level 2 | Level 3 | |||||||||||||
| December 31, 2022 | ||||||||||||||||
| Assets | ||||||||||||||||
| Investments held in MML Account | ||||||||||||||||
| Money Market Securities | $ | 259,392 | $ | 259,392 | $ | — | $ | — | ||||||||
| Certificate of Deposit | 100,823 | — | 100,823 | — | ||||||||||||
| December 31, 2021 | ||||||||||||||||
| Assets | ||||||||||||||||
| Investments held in MML Account | ||||||||||||||||
| Money Market Securities | $ | 300,051 | $ | 300,051 | $ | — | $ | — | ||||||||
| Certificate of Deposit | 100,722 | — | 100,722 | — | ||||||||||||
5. Property and Equipment, Net
Property and equipment, net, consist of the following as of December 31:
| 2022 | 2021 | |||||||
| Computer equipment | $ | 46,368 | $ | 19,631 | ||||
| Computer software | 244,990 | 244,990 | ||||||
| Equipment | 494,524 | 531,845 | ||||||
| Furniture and fixtures | 14,472 | 14,472 | ||||||
| Leasehold improvements | 66,985 | 76,405 | ||||||
| Vehicles | 239,565 | 174,338 | ||||||
| Property and equipment, gross | 1,106,904 | 1,061,681 | ||||||
| Less: accumulated depreciation | (778,043 | ) | (777,904 | ) | ||||
| Property and equipment, net | $ | 328,861 | $ | 283,777 | ||||
Depreciation expense for the years ended December 31, 2022 and 2021 were $99,162 and $91,426, respectively.
6. Accrued Expenses
The following is a summary of the Company’s accrued expenses as of December 31:
| 2022 | 2021 | |||||||
| Accrued payroll and payroll taxes | $ | 186,030 | $ | 172,757 | ||||
| Locums, temporary payroll | 128,337 | — | ||||||
| Data search fees | — | 19,651 | ||||||
| Billing fees | 34,510 | 19,293 | ||||||
| Other accrued expenses | 2,399 | 25,515 | ||||||
| $ | 351,276 | $ | 237,216 | |||||
| F-38 |
VILLAGE OAKS PATHOLOGY SERVICES, P.A.
D/B/A PRECISION PATHOLOGY SERVICES
NOTES TO FINANCIAL STATEMENTS
7. Leases
The Company has one operating lease for its real estate and office space and multiple finance leases for lab equipment in Texas. The operating lease has a remaining lease term of 4.58 years as of December 31, 2022. The Company has finance leases consisting of office and lab equipment with remaining lease terms ranging from approximately 0.9 to 5.0 years as of December 31, 2022, for which the Company has determined that it will use the equipment for a major part of its remaining economic life.
The lease agreements generally do not provide an implicit borrowing rate. Therefore, the Company used a benchmark approach as of January 1, 2022, to derive an appropriate incremental borrowing rate to discount remaining lease payments. The Company benchmarked itself against other companies of similar credit ratings and comparable quality and derived imputed rates ranging from 2.3% - 4.4% for lease term lengths ranging from approximately 1.9 to 5.6 years.
Leases with an initial term of twelve months or less are not recorded on the balance sheet. There are no material residual guarantees associated with any of the Company’s leases, and there are no significant restrictions or covenants included in the Company’s lease agreements. Certain leases include variable payments related to common area maintenance and property taxes, which are billed by the landlord, as is customary with these types of charges for office space. The Company has not entered into any lease arrangements with related parties, and the Company is not the sublessor in any arrangement.
The Company’s existing leases contain escalation clauses and renewal options. The Company has evaluated several factors in assessing whether there is reasonable certainty that the Company will exercise a contractual renewal option. For leases with renewal options that are reasonably certain to be exercised, the Company included the renewal term in the total lease term used in calculating the right-of-use asset and lease liability. Prior to adoption of ASU 2016-02 effective January 1, 2022, the Company accounted for operating lease transactions by recording lease expense on a straight-line basis over the expected term of the lease.
The components of lease expense, which are included in selling, general and administrative expense as of December 31, 2022 are as follows:
| Components of total lease expense: | 2022 | |||
| Amortization of ROU assets – finance lease | $ | 445,055 | ||
| Interest on lease liabilities – finance lease | 46,425 | |||
| Operating lease cost | 119,510 | |||
| Total lease cost | $ | 610,990 | ||
Supplemental balance sheet information relating to leases was as follows as of December 31, 2022:
| Operating leases: | 2022 | |||
| Operating lease right-of-use assets | $ | 494,900 | ||
| Operating lease liability, current | 96,654 | |||
| Operating lease liability, long-term | 403,177 | |||
| Finance leases: | 2022 | |||
| Finance lease right-of-use asset, gross | $ | 1,999,944 | ||
| Accumulated amortization | (445,055 | ) | ||
| Finance lease right-of-use asset, net | $ | 1,554,889 | ||
| Finance lease liability, current | $ | 413,729 | ||
| Finance lease liability, long-term | 1,218,535 | |||
| Total finance lease liability | $ | 1,632,264 | ||
| F-39 |
VILLAGE OAKS PATHOLOGY SERVICES, P.A.
D/B/A PRECISION PATHOLOGY SERVICES
NOTES TO FINANCIAL STATEMENTS
| Weighted-average remaining lease term: | 2022 | |||
| Operating leases (in years) | 4.58 | |||
| Finance leases (in years) | 4.07 | |||
| Weighted-average discount rate: | 2022 | |||
| Operating leases | 4.36 | % | ||
| Finance leases | 6.21 | % | ||
Future lease payments under non-cancelable operating leases as of December 31, 2022 were as follows:
| Year ending | Operating Leases | |||
| 2023 | $ | 116,498 | ||
| 2024 | 121,726 | |||
| 2025 | 121,726 | |||
| 2026 | 121,726 | |||
| 2027 and thereafter | 71,007 | |||
| Total Minimum Lease Payments | $ | 552,683 | ||
| Less effects of discounting | (52,852 | ) | ||
| Present value of future minimum lease payments | $ | 499,831 | ||
Future lease payments under non-cancelable finance leases as of December 31, 2022 were as follows:
| Year ending | Finance Leases | |||
| 2023 | $ | 505,266 | ||
| 2024 | 448,505 | |||
| 2025 | 448,505 | |||
| 2026 | 270,395 | |||
| 2027 and thereafter | 202,970 | |||
| Total Minimum Lease Payments | $ | 1,875,641 | ||
| Less effects of discounting | (243,377 | ) | ||
| Present value of future minimum lease payments | $ | 1,632,264 | ||
As described in Note 2, the Company adopted Topic 842 as of January 1, 2022. The prior year amounts have not been adjusted and continue to be reported in accordance with the Company’s historic accounting under Topic 840. The Company recognized lease expense of approximately $146,823 for the period ended December 31, 2021. There are no contingent rental amounts due to the lessors. Future minimum lease payments under non-cancellable leases as of December 31, 2021, were as follows:
| Year Ending December 31, | ||||
| 2022 | $ | 114,579 | ||
| 2023 | 116,498 | |||
| 2024 | 121,726 | |||
| 2025 | 121,726 | |||
| 2026 and thereafter | 192,733 | |||
| Total future minimum payments | $ | 667,262 | ||
8. Notes Payable
Hyundai Elantra – 2018
On May 10, 2022, the Company entered into a Finance Agreement to purchase a 2018 Hyundai Elantra for $19,444 with a maturity date of May 10, 2027. The loan bears fixed interest at a rate of 9.94% per annum, with monthly payments of $414, which is comprised of principal and interest. This loan is collateralized by the underlying vehicle. The balance of this loan as of December 31, 2022 and December 31, 2021 is $17,627 and $0, respectively.
| F-40 |
VILLAGE OAKS PATHOLOGY SERVICES, P.A.
D/B/A PRECISION PATHOLOGY SERVICES
NOTES TO FINANCIAL STATEMENTS
Hyundai Elantra – 2019
On May 10, 2022, the Company entered into a Finance Agreement to purchase a 2019 Hyundai Elantra for $20,509 with a maturity date of May 10, 2027. The loan bears fixed interest at a rate of 9.79% per annum, with monthly payments of $435, which is comprised of principal and interest. This loan is collateralized by the underlying vehicle. The balance of this loan as of December 31, 2022 and December 31, 2021 is $18,586 and $0, respectively.
Ford Transit – 2016
On May 4, 2016, the Company entered into a Finance Agreement to purchase a 2016 Ford Transit for $26,226 with a maturity date of June 4, 2022. The loan bears fixed interest at a rate of 5.39% per annum, with monthly payments of $428, which is comprised of principal and interest. This loan is collateralized by the underlying vehicle. The balance of this loan as of December 31, 2022 and December 31, 2021 is $0 and $2,530, respectively.
Hyundai Elantra - 2016
On March 4, 2021, the Company entered into a Finance Agreement to purchase a 2016 Hyundai Elantra for $13,609 with a maturity date of March 18, 2026. The loan bears fixed interest at a rate of 7.85% per annum, with monthly payments of $276, which is comprised of principal and interest. This loan is collateralized by the underlying vehicle. The balance of this loan as of December 31, 2022 and December 31, 2021 is $9,419 and $11,883, respectively.
Hyundai Elantra - 2017
On December 15, 2020, the Company entered into a Finance Agreement to purchase a 2017 Hyundai Elantra for $11,833 with a maturity date of December 15, 2024. The loan bears fixed interest at a rate of 9.84% per annum, with monthly payments of $300, which is comprised of principal and interest. This loan is collateralized by the underlying vehicle. The balance of this loan as of December 31, 2022 and December 31, 2021 is $6,462 and $9,279, respectively.
Hyundai Elantra - 2017
On December 15, 2020, the Company entered into a Finance Agreement to purchase a 2017 Hyundai Elantra for $10,000 with a maturity date of December 15, 2024. The loan bears fixed interest at a rate of 9.69% per annum, with monthly payments of $253, which is comprised of principal and interest. This loan is collateralized by the underlying vehicle. The balance of this loan as of December 31, 2022 and December 31, 2021 is $5,455 and $7,837, respectively.
Hyundai Elantra - 2020
On October 29, 2019, the Company entered into a Finance Agreement to purchase a 2020 Hyundai Elantra for $17,655 with a maturity date of October 29, 2024. The loan bears fixed interest at a rate of 7.24% per annum, with monthly payments of $352, which is comprised of principal and interest. This loan is collateralized by the underlying vehicle. The balance of this loan as of December 31, 2022 and December 31, 2021 is $7,090 and 10,655, respectively.
Hyundai Tucson
On August 28, 2020, the Company entered into a Finance Agreement to purchase a 2020 Hyundai Tucson for $24,841 with a maturity date of August 28, 2025. The loan has no stated interest rate and no effective interest rate with monthly principal payments of $414. This loan is collateralized by the underlying vehicle. The balance of this loan as of December 31, 2022 and December 31, 2021 is $13,249 and $18,217, respectively.
| F-41 |
VILLAGE OAKS PATHOLOGY SERVICES, P.A.
D/B/A PRECISION PATHOLOGY SERVICES
NOTES TO FINANCIAL STATEMENTS
Promissory Note - Fischer Equipment
On March 29, 2021, the Company entered into a $31,087 promissory note to finance the purchase of laboratory equipment. The promissory note bears interest at 4.25% per annum, with monthly payments of $577, which is comprised of principal and interest. This loan is collateralized by the underlying equipment. The balance of this note as of December 31, 2022 and December 31, 2021 is $20,928 and $26,840, respectively.
Promissory Note - BNB
On September 28, 2021, the Company entered into a $48,718 promissory note to finance the purchase of an Excellstoras Processor. The promissory bears fixed interest at 4.25% per annum, with monthly payments of $904, which is comprised of principal and interest. This loan is collateralized by the underlying equipment. The balance of this note as of December 31, 2022 and December 31, 2021 is $37,470 and $46,506, respectively.
Interest expense for all notes payable was $16,883 and $12,730 for the years ended December 31, 2022 and 2021, respectively.
Future minimum debt payments at December 31, 2022, are as follows:
| Years Ending December 31, | ||||
| 2023 | $ | 40,407 | ||
| 2024 | 40,523 | |||
| 2025 | 31,837 | |||
| 2026 | 19,631 | |||
| 2027 | 3,888 | |||
| Thereafter | - | |||
| Total | 136,286 | |||
| Less: Current Portion | (40,407 | ) | ||
| Notes payable, long-term | $ | 95,879 | ||
Line of Credit
On June 20, 2012, the Company entered into a Loan Agreement that provides the Company with a $200,000 revolving line of credit for the working capital needs of the Company with a maturity date of July 22, 2023. The Company may borrow, repay, and re-borrow at any time or from time to time while the line of credit is in effect. The line of credit was unsecured and not collateralized by any of the Company’s assets. Interest on the line of credit will accrue from the date of advance until final payment thereof at 1.00% above the prime rate. As of December 31, 2022 and 2021 there were no amounts outstanding under the line of credit.
9. Paycheck Protection Program
On April 17, 2020, the Company received $503,900 of proceeds under the Paycheck Protection Program (PPP) established pursuant to the CARES Act and administered by The Small Business Association (the “SBA”), as amended by the Paycheck Protection Program Flexibility Act of 2020 on June 22, 2020. The proceeds were recorded as debt, bear interest at 1% per annum and were unsecured. Amounts received under the PPP were used entirely to fund payroll costs as defined in the CARES Act and are expected to be eligible for forgiveness.
As of December 31, 2020, the Company used $503,900 of the loan proceeds to fund its payroll and related operational expenses. The Company submitted an application to the SBA on May 14, 2021, requesting these PPP funds received be forgiven. On September 9, 2021, the Company received notification the $503,900 was forgiven. As such, this amount has been recognized as other income in the accompanying statement of operations for the year ended December 31, 2021.
On March 30, 2021, the Company received an additional $503,950 under the Paycheck Protection Program (PPP) established pursuant to the CARES Act and administered by The Small Business Association (the “SBA”), as amended by the Paycheck Protection Program Flexibility Act of 2020 on June 22, 2020. The proceeds were recorded as debt, bear interest at 1% per annum and were unsecured. Amounts received under the PPP were used entirely to fund payroll costs as defined in the CARES Act and are expected to be eligible for forgiveness. As of December 31, 2021, the Company had not met the criteria for loan forgiveness. As such, the $503,950 of PPP funding is presented as long-term debt as of December 31, 2021. On April 4, 2022, the Company received notification the $503,950 was forgiven. As such, this amount has been recognized as other income in the accompanying statement of operations for the year ended December 31, 2022.
Based on current SBA guidance, the SBA has 6 years (up to 2026) to audit the good faith certification of eligibility and expenditures related to the Company’s PPP loan proceeds.
| F-42 |
VILLAGE OAKS PATHOLOGY SERVICES, P.A.
D/B/A PRECISION PATHOLOGY SERVICES
NOTES TO FINANCIAL STATEMENTS
10. Related Party Transactions
The majority shareholder of the Company is also an employee of the Company. Salaries paid to the majority shareholder for the years ended December 31, 2022 and 2021 were $590,000 and $597,000, respectively, and are included in selling, general, and administrative expenses in the accompanying statement of operations. The Company made distributions of $12,160 and $12,000 for the years ended December 31, 2022 and 2021, respectively to the majority shareholder.
11. Commitments and Contingencies
Litigation
From time to time, the Company may become subject to legal proceedings, claims or litigation arising in the ordinary course of business. In addition, the Company may receive notices alleging infringement of patents or other intellectual property rights. If an unfavorable outcome were to occur in litigation, the impact could be material to the Company’s business, financial condition, cash flow or results of operations, depending on the specific circumstances of the outcome. The Company accrues for loss contingencies when it is both probable that the Company will incur the loss and when it can reasonably estimate the amount of the loss or range of loss. As of December 31, 2022 and 2021, management believes there are no such outstanding claims or lawsuits that, individually or in the aggregate, would have a material adverse effect on the Company’s financial position, the results of its operations, or its cash flows.
bioAffinity Technologies, Inc. License Agreement
The Company has a license with bioAffinity Technologies, Inc. (“bioAffinity”) which allows the Company the use of bioAffinity’s proprietary CyPath® technology to provide patients with a diagnostic test for the detection of cancer. The license has an initial term through the date that the Company obtains FDA approval to directly commercialize similar equipment (or a functional equivalent of the licensed equipment). This license provides for certain royalties based on a percentage of services rendered. As of December 31, 2022 and 2021, there have been no payments made under the license agreement.
12. Retirement Plan
The Company maintains a 401(k) plan for qualified employees. The plan covers substantially all full-time employees of the Company who meet certain age and length of service requirements. There is no requirement for the Company to match employee contributions to the plan. The Company did not contribute to the plan during the years ended December 31, 2022 and 2021.
13. Subsequent Events
The Company has evaluated subsequent events occurring after the balance sheet date through the date of September 19, 2023, which is the date the financial statements were available to be issued. Based on this evaluation, the Company has determined the following subsequent events have occurred which require disclosure in the financial statements.
On September 18, 2023, the Company entered into an Asset Purchase Agreement (the “Asset Purchase Agreement”) wherein the Company was acquired by Precision Pathology Laboratory Services, LLC, a Texas limited liability company (“PPLS”), that is a wholly owned subsidiary of bioAffinity Technologies, Inc. (“bioAffinity”). Pursuant to the terms of the Asset Purchase Agreement the Company received $3,500,000 in consideration for the assets to be purchased by PPLS, of which $1,000,000 was paid by the issuance of 564,972 shares of bioAffinity’s restricted Common Stock to a trust controlled by Dr. Joyce (the “Joyce Trust”), which share number was based on the average of the trading day closing prices of bioAffinity for the 30 days prior to September 15, 2023, rounded to the nearest whole share.
| F-43 |
VILLAGE OAKS PATHOLOGY SERVICES, P.A.
D/B/A PRECISION PATHOLOGY SERVICES
Balance Sheets
| As of June 30, | As of December 31, | |||||||
| 2023 | 2022 | |||||||
| (unaudited) | ||||||||
| ASSETS | ||||||||
| Current Assets | ||||||||
| Cash | $ | 9,421 | $ | 357,470 | ||||
| Certificates of deposit | 100,823 | 100,823 | ||||||
| Investments | 272,404 | 259,392 | ||||||
| Patient fees receivable | 869,118 | 858,950 | ||||||
| Other receivables | 461,674 | 381,204 | ||||||
| Prepaid expenses | 9,316 | 31,123 | ||||||
| Total Current Assets | 1,722,756 | 1,988,962 | ||||||
| Non-Current Assets | ||||||||
| Property and equipment, net | 339,978 | 328,861 | ||||||
| Operating lease right-of-use asset, net | 445,599 | 494,900 | ||||||
| Finance lease right-of-use asset, net | 1,183,652 | 1,554,889 | ||||||
| Deposits | 8,000 | 8,000 | ||||||
| Total Non-Current Assets | 1,977,229 | 2,386,650 | ||||||
| TOTAL ASSETS | $ | 3,699,985 | $ | 4,375,612 | ||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY | ||||||||
| Current Liabilities | ||||||||
| Accounts payable | $ | 65,644 | $ | 83,386 | ||||
| Accrued expenses | 247,130 | 351,276 | ||||||
| Notes payable, current portion | 19,506 | 40,407 | ||||||
| Operating lease liability, current portion | 101,570 | 96,654 | ||||||
| Finance lease liability, current portion | 393,626 | 413,729 | ||||||
| Line of credit | 198,000 | — | ||||||
| Total Current Liabilities | 1,025,476 | 985,452 | ||||||
| Non-Current Liabilities | ||||||||
| Operating lease liability, net of current portion | 350,619 | 403,177 | ||||||
| Finance lease liability, net of current portion | 1,031,917 | 1,218,535 | ||||||
| Notes payable, net of current portion | 112,424 | 95,879 | ||||||
| Total Non-Current Liabilities | 1,494,960 | 1,717,591 | ||||||
| TOTAL LIABILITIES | 2,520,436 | 2,703,043 | ||||||
| Commitments and contingencies (see Note 11) | ||||||||
| Stockholders’ Equity | ||||||||
| Common stock, authorized 1,000, $0.01 par value; 500 shares issued and outstanding as of June 30, 2023 and December 31, 2022 | 5 | 5 | ||||||
| Retained earnings | 1,179,544 | 1,672,564 | ||||||
| Total Stockholders’ Equity | 1,179,549 | 1,672,569 | ||||||
| TOTAL LIABILITIES AND STOCKHOLDERS’ EQUITY | $ | 3,699,985 | $ | 4,375,612 | ||||
See accompanying notes to the unaudited condensed financial statements
| F-44 |
VILLAGE OAKS PATHOLOGY SERVICES, P.A.
D/B/A PRECISION PATHOLOGY SERVICES
Statements of Operations
(Unaudited)
| For the Six Months Ended June 30, | ||||||||
| 2023 | 2022 | |||||||
| Net Revenue | $ | 3,610,549 | $ | 3,230,545 | ||||
| Operating Expenses | ||||||||
| Selling, general, and administrative | 3,633,108 | 3,552,975 | ||||||
| Depreciation and amortization | 430,844 | 268,022 | ||||||
| Total Operating Expenses | 4,063,952 | 3,820,997 | ||||||
| Loss from Operations | (453,403 | ) | (590,452 | ) | ||||
| Other Income (Expense) | ||||||||
| PPP loan forgiveness | — | 503,950 | ||||||
| Other income, net | 5,148 | 7,688 | ||||||
| Interest expense | (57,777 | ) | (28,189 | ) | ||||
| Investment income | 4,881 | 4,249 | ||||||
| Unrealized gain (loss) on investments | 8,131 | (44,507 | ) | |||||
| Total Other (Expense) Income | (39,617 | ) | 443,191 | |||||
| Net loss | $ | (493,020 | ) | $ | (147,261 | ) | ||
See accompanying notes to the unaudited condensed financial statements
| F-45 |
VILLAGE OAKS PATHOLOGY SERVICES, P.A.
D/B/A PRECISION PATHOLOGY SERVICES
Statements of Stockholders’ Equity
(Unaudited)
| Retained Earnings | Common Stock |
Total Stockholders’ Equity |
||||||||||
| Balance as of December 31, 2021 | $ | 2,146,356 | $ | 5 | $ | 2,146,361 | ||||||
| Distributions | (12,160 | ) | — | (12,160 | ) | |||||||
| Net loss | (147,261 | ) | — | (147,261 | ) | |||||||
| Balance as of June 30, 2022 | $ | 1,986,935 | $ | 5 | $ | 1,986,940 | ||||||
| Balance as of December 31, 2022 | $ | 1,672,564 | $ | 5 | $ | 1,672,569 | ||||||
| Net loss | (493,020 | ) | — | — | ||||||||
| Balance as of June 30, 2023 | $ | 1,179,544 | $ | 5 | $ | 1,179,549 | ||||||
See accompanying notes to the unaudited condensed financial statements
| F-46 |
VILLAGE OAKS PATHOLOGY SERVICES, P.A.
D/B/A PRECISION PATHOLOGY SERVICES
Statements of Cash Flows
(Unaudited)
| For the Six Months Ended June 30, | ||||||||
| 2023 | 2022 | |||||||
| Cash flows from operating activities: | ||||||||
| Net loss | $ | (493,020 | ) | $ | (147,261 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Forgiveness of PPP loan payable | — | (503,950 | ) | |||||
| Depreciation | 59,608 | 45,495 | ||||||
| Amortization of right-of-use asset | 371,236 | 222,527 | ||||||
| Gain on disposal of fixed assets | (4,801 | ) | — | |||||
| Investment income | (4,881 |
) | (4,249 | ) | ||||
| Unrealized (gain) loss on investments | (8,131 | ) | 44,507 | |||||
| Change in operating assets and liabilities: | ||||||||
| Patient fees receivable | (10,168 | ) | (95,403 | ) | ||||
| Other receivables | (80,470 | ) | 205,666 | |||||
| Prepaid expenses | 21,807 | (25,126 | ) | |||||
| Accounts payable | (17,742 | ) | (15,030 | ) | ||||
| Accrued expenses | (104,146 | ) | 55,365 | |||||
| Operating lease right-of-use asset | 1,659 | (11,037 | ) | |||||
| Net cash used in operating activities | (269,049 | ) | (228,496 | ) | ||||
| Cash flows from investing activities: | ||||||||
| Purchase of property and equipment | (76,642 | ) | (101,175 | ) | ||||
| Proceeds from disposals of property and equipment | 10,718 | — | ||||||
| Net cash used in investing activities | (65,924 | ) | (101,175 | ) | ||||
| Cash flows from financing activities: | ||||||||
| Borrowings on line of credit | 198,000 | — | ||||||
| Borrowings of notes payable | 20,210 | 39,953 | ||||||
| Repayments of notes payable | (24,566 | ) | (39,040 | ) | ||||
| Principal repayments on finance leases | (206,720 | ) | (182,295 | ) | ||||
| Distributions | — | (12,160 | ) | |||||
| Net cash used in financing activities | (13,076 | ) | (193,542 | ) | ||||
| Net decrease in cash | (348,049 | ) | (523,213 | ) | ||||
| Cash, beginning of year | 357,470 | 1,207,341 | ||||||
| Cash, end of year | $ | 9,421 | $ | 684,128 | ||||
See accompanying notes to the unaudited condensed financial statements
| F-47 |
VILLAGE OAKS PATHOLOGY SERVICES, P.A.
D/B/A PRECISION PATHOLOGY SERVICES
Statements of Cash Flows (Continued)
(Unaudited)
| 2023 | 2022 | |||||||
| Supplemental disclosure of cash flow information: | ||||||||
| Cash paid for interest | $ | 57,777 | $ | 28,189 | ||||
| Non-Cash Investing and Financing Transactions: | ||||||||
| Operating right-of-use asset obtained in exchange for lease liabilities | $ | — | $ | 590,474 | ||||
See accompanying notes to the unaudited condensed financial statements
| F-48 |
VILLAGE OAKS PATHOLOGY P.A.
D/B/A PRECISION PATHOLOGY SERVICES
NOTES TO UNAUDITED FINANCIAL STATEMENTS
Note 1 - Nature of Operations
Village Oaks Pathology Services, P.A., doing business as Precision Pathology Services (the “Company” or “Precision Pathology”) is a privately held company organized in 1987 under the laws of the state of Texas. Precision Pathology provides anatomic and clinical pathology services for patients and their physicians.
Income Taxes
The Company, with stockholders’ consent, has elected to be taxed as an “S Corporation” under the provisions of the Internal Revenue Code and comparable state income tax law. As an S Corporation, the Company is generally not subject to corporate income taxes and the Company’s net income or loss is reported on the individual tax return of the stockholders of the Company. Therefore, no provision or liability for income taxes is reflected in the financial statements. The Company has not been audited by the Internal Revenue Service, and accordingly the business tax returns since 2020 are open to examination. Management has evaluated its tax positions and has concluded that the Company had taken no uncertain tax positions that could require adjustment or disclosure in the financial statements to comply with provisions set forth in Accounting Standards Codification (“ASC”) Topic 740, Income Taxes.
Note 2 - Summary of Significant Accounting Policies
Basis of Presentation
The accompanying unaudited interim financial statements have been prepared in accordance with generally accepted accounting principles in the United States of America (“GAAP”). Accordingly, they do not include certain footnotes and financial presentations normally required under GAAP for complete financial statements.
These unaudited condensed financial statements should be read in conjunction with the Company’s audited financial statements for the years ended December 31, 2022 and 2021 that were issued on September 19, 2023. In management’s opinion, the accompanying unaudited condensed financial statements contain all adjustments consisting of normal, recurring and non-recurring adjustments that were considered necessary for the fair presentation of the Company’s financial position, results of operations, and cash flows as of the dates and for the periods presented.
Use of Estimates
The preparation of financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. The Company evaluates estimates and assumptions on a regular basis. The Company bases its estimates and assumptions on current facts, historical experience and various other factors that it believes to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities and the accrual of costs and expenses that are not readily apparent from other sources. The Company’s accounting policies that involve significant judgment and estimates include revenue recognition including contractual adjustments and discounts, patient fee receivables and the related allowance for contractual discounts and allowance for doubtful accounts, valuation of the lease liabilities and related right-of-use-assets, and estimates of useful lives for depreciation. The actual results experienced by the Company may differ materially and adversely from the Company’s estimates. To the extent there are material differences between the estimates and the actual results, future results of operations will be affected.
| F-49 |
VILLAGE OAKS PATHOLOGY P.A.
D/B/A PRECISION PATHOLOGY SERVICES
NOTES TO UNAUDITED FINANCIAL STATEMENTS
Fair Value Measurements
The Company applies Accounting Standards Codification (“ASC”) Topic 820, Fair Value Measurement (“ASC 820”), which establishes a framework for measuring fair value and clarifies the definition of fair value within that framework. ASC 820 defines fair value as an exit price, which is the price that would be received for an asset or paid to transfer a liability in the Company’s principal or most advantageous market in an orderly transaction between market participants on the measurement date. The fair value hierarchy established in ASC 820 generally requires an entity to maximize the use of observable inputs and minimize the use of unobservable inputs when measuring fair value. Observable inputs reflect the assumptions that market participants would use in pricing the asset or liability and are developed based on market data obtained from sources independent of the reporting entity. Unobservable inputs reflect the entity’s own assumptions based on market data and the entity’s judgments about the assumptions that market participants would use in pricing the asset or liability and are to be developed based on the best information available in the circumstances.
The carrying amounts reflected in the balance sheet for current assets and liabilities approximate fair value due to their short-term nature.
| Level 1 — Assets and liabilities with unadjusted, quoted prices listed on active market exchanges. Inputs to the fair value measurement are observable inputs, such as quoted prices in active markets for identical assets or liabilities. | |
| Level 2 — Inputs to the fair value measurement are determined using prices for recently traded assets and liabilities with similar underlying terms, as well as direct or indirect observable inputs, such as interest rates and yield curves that are observable at commonly quoted intervals. | |
| Level 3 — Inputs to the fair value measurement are unobservable inputs, such as estimates, assumptions, and valuation techniques when little or no market data exists for the assets or liabilities. |
See Note 4 for additional information on assets measured at fair value.
Liquidity and Capital Resources
In accordance with Accounting Standards Update (“ASU”) 2014-15, Presentation of Financial Statements – Going Concern (Subtopic 205-40), the Company has evaluated whether there are conditions and events that raise substantial doubt about the Company’s ability to continue as a going concern for at least one year after the date the condensed financial statements are issued. As required by this standard, management’s evaluation shall initially not take into consideration the potential mitigating effects of management’s plans that have not been fully implemented as of the date the financial statements are issued.
The Company’s assessment included the preparation of a detailed cash forecast that included all projected cash inflows and outflows. Although the Company continues to focus on growing its revenues, the Company’s ongoing operating expenditures will exceed the revenue it expects to receive for the foreseeable future. Additionally, the Company has a history of operating losses and negative operating cash flows and expects these trends to continue. Our future plans may include cash flows generated from our revenues, issuance of debt, or sales of our equity securities.
The Company’s loss from operations before depreciation and amortization was ($22,559) for the six months ended June 30, 2023. Cash used for operating activities and financing debt payments for the six months ended June 30, 2023 was ($269,049) and ($231,286), respectively. The Company’s cash, certificates of deposit and investments on hand as of June 30, 2023, was $382,648. Based on the cash on hand and current projections of cash requirements from operating, investing, and financing activities, management concludes that there is substantial doubt about the Company’s ability to continue as a going concern without putting in place a mitigating plan or raising additional funds through debt or capital for purposes of issuing the interim financials for the six-month period ended June 30, 2023. Despite a history of successfully implementing similar plans to alleviate adverse financial conditions, these sources of working capital are not currently assured, and consequently do not sufficiently mitigate the risks and uncertainties disclosed above. These unaudited condensed financial statements do not include any adjustments to reflect the possible future effects on the recoverability and classification of assets or the amounts of liabilities that may result from uncertainty related to the Company’s ability to continue as a going concern.
| F-50 |
VILLAGE OAKS PATHOLOGY P.A.
D/B/A PRECISION PATHOLOGY SERVICES
NOTES TO UNAUDITED FINANCIAL STATEMENTS
Cash
The Company’s cash is held with one financial institution, and the account balances may exceed the Federal Deposit Insurance Corporation (“FDIC”) insurance limit at times. Accounts are insured by the FDIC up to $250,000. As of June 30, 2023 and December 31, 2022, the Company had uninsured cash deposits of $0 and $107,470, respectively. The Company has not experienced any losses in such accounts to date. Any loss incurred or a lack of access to such funds could have a significant adverse impact on the Company’s financial condition, results of operations, and cash flows. All highly liquid investments with maturities of three months or less at the date of purchase are classified as cash equivalents.
Investments held in MML Investors Services Account
As of June 30, 2023 and December 31, 2022, the assets held in the MML Investors Services Account (“MML Account”) were held in money market funds, which are invested in fixed income and equity securities. Trading securities are presented on the balance sheet at fair value at the end of each reporting period. Gains and losses resulting from the change in fair value of these securities is included in unrealized losses on investments in the accompanying statement of operations. Dividend income and short-term and long-term capital gains on these securities is included in investment income in the accompanying statement of operations. As of June 30, 2023 and December 31, 2022, the assets held in the MML Account were $272,404 and $259,392, respectively.
Certificates of Deposit
The Company invests its excess cash in bank certificates of deposit (“CDs”) which are fully insured by the FDIC with terms of not more than six months. As of June 30, 2023 and December 31, 2022, the Company had certificates of deposit with balances of $100,823 and $100,823, respectively.
Patient Fees Receivable
Patient accounts receivable represents amounts due from patient services billed to commercial insurance companies, governmental payors, and patients. Receivables are recorded at the amount the Company expects to collect. The Company estimates variable consideration for patient service fees using an expected value method. Accordingly, the Company has developed ratios for portfolios of payors based on the nature of the payor (e.g., commercial insurer, government program, uninsured patients), which impacts the average time to collect the consideration to which the Company expects to be entitled and the amount of such consideration. The Company has developed payment-to-charge ratio for each portfolio of payor based on historical payment experience and applied those ratios to gross charges for each year presented in order to arrive at the net patient fees receivable.
Other Receivables
Other receivables represent amounts billed for pathologist interpretations and medical director fees, which include the Company’s pathologists providing directorship for certain hospital facilities. Other receivables are recorded at the amount the Company expects to collect. Management determines the allowance for credit losses based on historical collection experience, contract terms, and general and market business conditions. As of June 30, 2023 and December 31, 2022, management determined no allowance was necessary related to these receivables.
Property and Equipment
In accordance with ASC 360-10, Accounting for the Impairment of Long-Lived Assets, the Company periodically reviews the carrying value of its long-lived assets, such as property and equipment, to test whether current events or circumstances indicate that such carrying value may not be recoverable. When evaluating assets for potential impairment, the Company compares the carrying value of the asset to its estimated undiscounted future cash flows. If an asset’s carrying value exceeds such estimated cash flows (undiscounted and with interest charges), the Company records an impairment charge for the difference. The Company did not record impairment for the six months ended June 30, 2023 and 2022.
| F-51 |
VILLAGE OAKS PATHOLOGY P.A.
D/B/A PRECISION PATHOLOGY SERVICES
NOTES TO UNAUDITED FINANCIAL STATEMENTS
Property and equipment are carried at cost, net of accumulated depreciation. Depreciation is computed using the straight-line method over the estimated useful life of the asset. Amortization of leasehold improvements is computed using the shorter of the lease term or estimated useful life of the asset. Additions and improvements are capitalized, while repairs and maintenance are expensed as incurred. Useful lives of each asset class are as follows:
| Asset Category | Useful Life | |
| Computer equipment | 5 years | |
| Computer software | 3 years | |
| Equipment | 5-7 years | |
| Furniture and fixtures | 5-7 years | |
| Vehicles | 5 years | |
| Leasehold improvements | Lesser of lease term or useful life |
Revenue Recognition
The Company derives revenues from providing pathology testing services to patients and other customers. Revenue from services is recognized upon the transfer of control, which is generally achieved when testing is completed and the results are delivered to a patient, a patient’s physician, or institutional customers such as independent laboratories, hospitals or contract research organizations (“CRO”). The Company’s revenues fall into three separate streams: (a) patient service fees, (b) histology service fees, and (c) medical director fees.
On January 1, 2021, the Company adopted ASC 606, Revenue from Contracts with Customers (“ASC 606”), using the modified retrospective method with respect to all non-completed contracts. ASC 606 outlines a single comprehensive model for entities to use in accounting for revenue arising from contracts with customers and supersedes nearly all existing revenue recognition guidance, including industry-specific guidance.
The new guidance is based on the principle that an entity should recognize revenue to depict the transfer of products or services to customers in an amount that reflects the consideration to which the entity expects to be entitled in exchange for those products or services. The adoption of ASC 606 did not have a material effect on the Company’s financial position, results of operations, or internal controls over financial reporting.
The Company determines revenue recognition by applying the following steps prescribed under ASC 606:
| a. | Identification of the contract, or contracts, with a customer; | |
| b. | Identification of the performance obligations in the contract; | |
| c. | Determination of the transaction price; | |
| d. | Allocation of the transaction price to the performance obligations in the contract; and | |
| e. | Recognition of revenue when, or as, we satisfy a performance obligation. |
The Company collects patient service fees from patients and various third-party payors, mainly insurance companies and governmental payors. Patient service fees are earned from performing pathology lab services (procedures or tests), which may be requested by a patient directly or by a physician on a patient’s behalf. The Company also provides histology services to hospitals, CRO’s or independent laboratories. The Company’s services represent performance obligations transferred to the customer at the point in time when the test results are delivered, which is when the customer obtains the benefits of the service.
| F-52 |
VILLAGE OAKS PATHOLOGY P.A.
D/B/A PRECISION PATHOLOGY SERVICES
NOTES TO UNAUDITED FINANCIAL STATEMENTS
Patient service fee revenue is variable given various factors that impact whether third-party payors ultimately pay the Company’s contractual billing rates. While third-party payor rates are known at inception of the contract, the payor has the ultimate discretion to adjudicate claims and decide on the final payment amount. There are various factors that allow third-party payors the right to deny all or part of a claim, which may not be known at inception of the contract. While the Company may appeal claim denials or adjustments, generally the Company offers some level of implicit price concession as part of these adjustments made by payors. Furthermore, patient service fees billed to uninsured patients is subject to variability for factors not known at inception. In contrast, the transaction price for histology services is generally fixed, as no third-party payors are involved, and therefore, the fees agreed upon upfront are the fees that the Company expects to collect for services performed.
The Company estimates variable consideration for patient service fees using an expected value method. Accordingly, the Company has developed ratios for portfolios of payors based on the nature of the payor (e.g., commercial insurer, government program, uninsured patients), which impacts the average time to collect the consideration to which the Company expects to be entitled and the amount of such consideration. The Company has developed payment-to-charge ratio for each portfolio of payors based on historical payment experience and applied those ratios to gross charges for each year presented. Variable consideration is constrained to the extent that it is deemed probable that a significant reversal in the amount of revenue recognized will not occur when the uncertainty is resolved, which is when an insurance claim is fully resolved.
Advertising
Advertising costs are expensed as incurred. Advertising costs were $1,645 and $2,645 for the six months ended June 30, 2023 and 2022, respectively, which are included in selling, general and administrative expense on the accompanying statements of operations.
Leases
The Company determines if an arrangement is a lease at inception and classifies its leases at commencement. Operating leases are presented as right-of-use (“ROU”) assets and the corresponding lease liabilities are included in operating lease liabilities, current and operating lease liabilities on the Company’s balance sheets. ROU assets represent the Company’s right to use an underlying asset, and lease liabilities represent the Company’s obligation for lease payments in exchange for the ability to use the asset for the duration of the lease term.
ROU assets and lease liabilities are recognized at commencement date and determined using the present value of the future minimum lease payments over the lease term. The Company used a discount rate based on a benchmark approach as of January 1, 2022, the date of initial application of the new guidance, to derive an appropriate incremental borrowing rate to discount remaining lease payments. The Company benchmarked itself against other companies of similar credit ratings and comparable quality and derived imputed rates for lease term lengths ranging from approximately 1.9 to 5.6 years. The lease term may include options to extend when it is reasonably certain that the Company will exercise that option. In addition, the Company does not recognize short-term leases that have a term of twelve months or less as ROU assets or lease liabilities. The Company recognizes operating lease expense on a straight-line basis over the lease term.
| F-53 |
VILLAGE OAKS PATHOLOGY P.A.
D/B/A PRECISION PATHOLOGY SERVICES
NOTES TO UNAUDITED FINANCIAL STATEMENTS
The Company has lease agreements which contain both lease and non-lease components, which it has elected to account for as a single lease component when the payments are fixed. As such, variable lease payments, including those not dependent on an index or rate, such as real estate taxes, common area maintenance, and other costs that are subject to fluctuation from period to period are not included in lease measurement.
Recent Accounting Pronouncements
In November 2018, the FASB issued ASU 2018-19, Codification Improvements to Topic 326, Financial Instruments—Credit Losses, which amends the guidance for accounting for assets that are potentially subject to credit risk. The amendments affect contract assets, loans, debt securities, trade receivables, net investments in leases, off-balance-sheet credit exposures, reinsurance receivables, and any other financial assets not excluded from the scope that have the contractual right to receive cash. ASU 2018-19 is effective for fiscal years beginning after December 15, 2022. The Company adopted ASU 2018-19 effective January 1, 2023 and the adoption of this guidance did not have a material impact on the Company’s condensed financial statements.
Note 3 – Revenue, net
The following is a summary of net revenue for the six months ended June 30:
2023 |
2022 |
|||||||
| Patient fees | $ | 2,890,746 | $ | 2,494,160 | ||||
| Histology service fees | 643,733 | 653,886 | ||||||
| Medical director fees | 37,201 | 57,801 | ||||||
| Other revenue | 38,869 | 24,698 | ||||||
| Revenue, net | $ | 3,610,549 | $ | 3,230,545 | ||||
Concentrations
The Company has contracts with various third-party payors, mainly insurance companies and governmental payors. There is no concentration of revenues from individual payor’s. For the six months ended June 30, 2023 and 2022, approximately 92% of revenues relate to contracts with commercial payors.
| F-54 |
VILLAGE OAKS PATHOLOGY SERVICES, P.A.
D/B/A PRECISION PATHOLOGY SERVICES
NOTES TO UNAUDITED FINANCIAL STATEMENTS
Note 4 - Investments
The following table presents information about the Company’s financial assets and liabilities that are measured at fair value as of June 30, 2023 and December 31, 2022, and indicates the fair value hierarchy of the valuation inputs the Company utilized to determine such fair value:
| Description | Fair Value | Level 1 | Level 2 | Level 3 | ||||||||||||
| June 30, 2023 | ||||||||||||||||
| Assets | ||||||||||||||||
| Investments held in MML Account | ||||||||||||||||
| Money Market Securities | $ | 272,404 | $ | 272,404 | $ | — | $ | — | ||||||||
| Certificate of Deposit | $ | 100,823 | $ | — | $ | 100,823 | $ | — | ||||||||
| December 31, 2022 | ||||||||||||||||
| Assets | ||||||||||||||||
| Investments held in MML Account | ||||||||||||||||
| Money Market Securities | $ | 259,392 | $ | 259,392 | $ | — | $ | — | ||||||||
| Certificate of Deposit | $ | 100,823 | $ | — | $ | 100,823 | $ | — | ||||||||
Note 5 - Property and Equipment, net
Property and equipment, net, consist of the following as of June 30, 2023 and December 31, 2022:
2023 |
2022 | |||||||
| Computer equipment | $ | 46,368 | $ | 46,368 | ||||
| Computer software | 244,990 | 244,990 | ||||||
| Equipment | 547,456 | 494,524 | ||||||
| Furniture and fixtures | 14,472 | 14,472 | ||||||
| Leasehold improvements | 66,985 | 66,985 | ||||||
| Vehicles | 247,942 | 239,565 | ||||||
| Property and equipment, gross | 1,168,213 | 1,106,904 | ||||||
| Less: accumulated depreciation | (828,235 | ) | (778,043 | ) | ||||
| Property and equipment, net | $ | 339,978 | $ | 328,861 | ||||
Depreciation expense for the six months ended June 30, 2023 and 2022 were $59,608 and $45,495, respectively.
| F-55 |
VILLAGE OAKS PATHOLOGY SERVICES, P.A.
D/B/A PRECISION PATHOLOGY SERVICES
NOTES TO UNAUDITED FINANCIAL STATEMENTS
Note 6 - Accrued Expenses
The following is a summary of the Company’s accrued expenses as of June 30, 2023 and December 31, 2022:
2023 |
2022 | |||||||
| Accrued payroll and payroll taxes | $ | 143,604 | $ | 186,030 | ||||
| Contractors | 63,000 | 128,337 | ||||||
| Billing fees | 37,163 | 34,510 | ||||||
| Other accrued expenses | 3,363 | 2,399 | ||||||
| $ | 247,130 | $ | 351,276 | |||||
Note 7 - Leases
The Company has one operating lease for its real estate and office space and multiple finance leases for lab equipment in Texas. The operating lease has a remaining lease term of 4.08 years as of June 30, 2023. The Company has finance leases consisting of office and lab equipment with remaining lease terms ranging from approximately 0.9 to 4.5 years as of June 30, 2023, for which the Company has determined that it will use the equipment for a major part of its remaining economic life.
The lease agreements generally do not provide an implicit borrowing rate. Therefore, the Company used a benchmark approach as of January 1, 2022, to derive an appropriate incremental borrowing rate to discount remaining lease payments. The Company benchmarked itself against other companies of similar credit ratings and comparable quality and derived imputed rates ranging from 2.3% - 4.4% for lease term lengths ranging from approximately 1.9 to 5.6 years.
Leases with an initial term of twelve months or less are not recorded on the balance sheet. There are no material residual guarantees associated with any of the Company’s leases, and there are no significant restrictions or covenants included in the Company’s lease agreements. Certain leases include variable payments related to common area maintenance and property taxes, which are billed by the landlord, as is customary with these types of charges for office space. The Company has not entered into any lease arrangements with related parties, and the Company is not the sublessor in any arrangement.
The Company’s existing leases contain escalation clauses and renewal options. The Company has evaluated several factors in assessing whether there is reasonable certainty that the Company will exercise a contractual renewal option. For leases with renewal options that are reasonably certain to be exercised, the Company included the renewal term in the total lease term used in calculating the right-of-use asset and lease liability. Prior to adoption of ASU 2016-02 effective January 1, 2022, the Company accounted for operating lease transactions by recording lease expense on a straight-line basis over the expected term of the lease.
| F-56 |
VILLAGE OAKS PATHOLOGY SERVICES, P.A.
D/B/A PRECISION PATHOLOGY SERVICES
NOTES TO UNAUDITED FINANCIAL STATEMENTS
The components of lease expense, which are included in selling, general and administrative expense for the six months ended June 30, 2023 and 2022 are as follows:
| Components of lease expense: | 2023 |
2022 |
||||||
| Amortization of ROU assets - finance lease | $ | 371,236 | $ | 222,527 | ||||
| Interest on lease liabilities - finance lease | 53,784 | 24,690 | ||||||
| Operating lease cost | 59,755 | 59,755 | ||||||
| Total lease cost | $ | 484,775 | $ | 306,972 | ||||
Supplemental balance sheet information relating to leases was as follows as of June 30, 2023 and December 31, 2022:
| Operating leases: | 2023 |
2022 | ||||||
| Operating lease right-of-use assets | $ | 445,599 | $ | 494,900 | ||||
| Operating lease liability, current | $ | 101,570 | $ | 96,654 | ||||
| Operating lease liability, long-term | $ | 350,619 | $ | 403,177 | ||||
| Finance leases: | 2023 |
2022 | ||||||
| Finance lease right-of-use asset, gross | $ | 1,999,944 | $ | 1,999,944 | ||||
| Accumulated amortization | (816,292 | ) | (445,055 | ) | ||||
| Finance lease right-of-use asset, net | 1,183,652 | 1,554,889 | ||||||
| Finance lease liability, current | $ | 393,626 | $ | 413,729 | ||||
| Finance lease liability, long-term | 1,031,917 | 1,218,535 | ||||||
| Total finance lease liabilities | $ | 1,425,543 | $ | 1,632,264 | ||||
| Weighted-average remaining lease term: | 2023 |
2022 | ||||||
| Operating leases (in years) | 4.08 | 4.58 | ||||||
| Finance leases (in years) | 3.65 | 4.07 | ||||||
| Weighted-average discount rate: | 2023 |
2022 | ||||||
| Operating leases | 4.36 | % | 4.36 | % | ||||
| Finance leases | 6.21 | % | 6.21 | % | ||||
| F-57 |
VILLAGE OAKS PATHOLOGY SERVICES, P.A.
D/B/A PRECISION PATHOLOGY SERVICES
NOTES TO UNAUDITED FINANCIAL STATEMENTS
Future minimum lease payments under non-cancellable lease as of June 30, 2023, are as follows:
| Year Ending December 31, | Operating Leases |
Finance Leases |
||||||
| Remaining 2023 | $ | 68,085 | $ | 250,053 | ||||
| 2024 | 121,726 | 448,505 | ||||||
| 2025 | 121,726 | 448,505 | ||||||
| 2026 | 121,726 | 270,395 | ||||||
| 2027 and thereafter | 71,007 | 202,970 | ||||||
| Total undiscounted cash flows | 504,270 | 1,620,428 | ||||||
| Less discounting | (52,081 | ) | (194,885 | ) | ||||
| Present value of lease liabilities | $ | 452,189 | $ | 1,425,543 | ||||
Note 8 - Notes Payable
Hyundai Elantra - 2020
On October 29, 2019, the Company entered into a Finance Agreement to purchase a 2020 Hyundai Elantra for $17,655 with a maturity date of October 29, 2024. The loan bears fixed interest at a rate of 7.24% per annum, with monthly payments of $352, which is comprised of principal and interest. This loan is collateralized by the underlying vehicle. The balance of this loan as of June 30, 2023 and December 31, 2022 is $5,214 and $7,090, respectively.
Hyundai Tucson - 2020
On August 28, 2020, the Company entered into a Finance Agreement to purchase a 2020 Hyundai Tucson for $24,841 with a maturity date of August 28, 2025. The loan has no stated interest rate with monthly principal payments of $414. This loan is collateralized by the underlying vehicle. The balance of this loan as of June 30, 2023 and December 31, 2022 is $10,765 and $13,249, respectively.
Hyundai Elantra – 2017
On December 15, 2020, the Company entered into a Finance Agreement to purchase a 2017 Hyundai Elantra for $10,000 with a maturity date of December 15, 2024. The loan bears fixed interest at a rate of 9.69% per annum, with monthly payments of $253, which is comprised of principal and interest. This loan is collateralized by the underlying vehicle. The balance of this loan as of June 30, 2023 and December 31, 2022 is $4,176 and $5,455, respectively.
Hyundai Elantra - 2016
On March 4, 2021, the Company entered into a Finance Agreement to purchase a 2016 Hyundai Elantra for $13,609 with a maturity date of March 18, 2026. The loan bears fixed interest at a rate of 7.85% per annum, with monthly payments of $276, which is comprised of principal and interest. This loan is collateralized by the underlying vehicle. The balance of this loan as of June 30, 2023 and December 31, 2022 is $8,052 and $9,419, respectively.
| F-58 |
VILLAGE OAKS PATHOLOGY SERVICES, P.A.
D/B/A PRECISION PATHOLOGY SERVICES
NOTES TO UNAUDITED FINANCIAL STATEMENTS
Promissory Note - Fischer Equipment
On March 29, 2021, the Company entered into a $31,087 promissory note to finance the purchase of laboratory equipment. The promissory note bears interest at 4.25% per annum, with monthly payments of $577, which is comprised of principal and interest. This loan is collateralized by the underlying equipment. The balance of this note as of June 30, 2023 and December 31, 2022 is $17,913 and $20,928, respectively.
Promissory Note - BNB
On September 28, 2021, the Company entered into a $48,718 promissory note to finance the purchase of an Excellstoras Processor. The promissory bears fixed interest at 4.25% per annum, with monthly payments of $904, which is comprised of principal and interest. This loan is collateralized by the underlying equipment. The balance of this note as of June 30, 2023 and December 31, 2022 is $32,819 and $37,470, respectively.
Hyundai Elantra - 2018
On May 10, 2022, the Company entered into a Finance Agreement to purchase a 2018 Hyundai Elantra for $19,444 with a maturity date of May 10, 2027. The loan bears fixed interest at a rate of 9.94% per annum, with monthly payments of $414, which is comprised of principal and interest. This loan is collateralized by the underlying vehicle. The balance of this loan as of June 30, 2023 and June 30, 2022 is $15,986 and $17,627, respectively.
Hyundai Elantra - 2019
On May 10, 2022, the Company entered into a Finance Agreement to purchase a 2019 Hyundai Elantra for $20,509 with a maturity date of May 10, 2027. The loan bears fixed interest at a rate of 9.79% per annum, with monthly payments of $435, which is comprised of principal and interest. This loan is collateralized by the underlying vehicle. The balance of this loan as of June 30, 2023 and December 31, 2022 is $16,853 and $18,586, respectively.
Hyundai Elantra - 2017
On December 15, 2020, the Company entered into a Finance Agreement to purchase a 2017 Hyundai Elantra for $11,833 with a maturity date of December 15, 2024. The loan bears fixed interest at a rate of 9.84% per annum, with monthly payments of $300, which is comprised of principal and interest. This loan is collateralized by the underlying vehicle. The balance of this loan as of June 30, 2023 and December 31, 2022 is $0 and $6,462, respectively, as the Company paid the loan off early.
Hyundai Elantra - 2018
On June 27, 2023, the Company entered into a Finance Agreement to purchase a 2018 Hyundai Elantra for $20,210 with a maturity date of July 27, 2029. The loan bears fixed interest at a rate of 10.64% per annum, with monthly payments of $383, which is comprised of principal and interest. This loan is collateralized by the underlying vehicle. The balance of this loan as of June 30, 2023 and December 31, 2022 is $20,152 and $0, respectively.
Interest expense for all notes payable was $3,993 and $3,499 for the six months ended June 30, 2023 and 2022, respectively.
| F-59 |
VILLAGE OAKS PATHOLOGY SERVICES, P.A.
D/B/A PRECISION PATHOLOGY SERVICES
NOTES TO UNAUDITED FINANCIAL STATEMENTS
Future minimum debt payments at June 30, 2023, are as follows:
| Year Ending December 31, | ||||
| Remaining 2023 | $ | 19,506 | ||
| 2024 | 40,523 | |||
| 2025 | 34,804 | |||
| 2026 | 22,931 | |||
| 2027 | 7,559 | |||
| Thereafter | 6,607 | |||
| Total | 131,930 | |||
| Less: current portion | (19,506 | ) | ||
| $ | 112,424 | |||
Line of Credit
On June 20, 2012, the Company entered into a Loan Agreement that provides the Company with a $200,000 revolving line of credit for the working capital needs of the Company with a maturity date of July 22, 2023. The Company may borrow, repay, and re-borrow at any time or from time to time while the line of credit is in effect. The line of credit was unsecured and not collateralized by any of the Company’s assets.
Interest on the line of credit will accrue from the date of advance until final payment thereof at 1.00% above the prime rate (9.25% as of June 30, 2023). As of June 30, 2023 and December 31, 2022 the Company had $198,000 and $0 outstanding on the line of credit, respectively.
Note 9 - Provider Relief Funds and Paycheck Protection Program
Paycheck Protection Program
On March 30, 2021, the Company received $503,950 under the Paycheck Protection Program (“PPP”) established pursuant to the CARES Act and administered by The Small Business Association (the “SBA”), as amended by the Paycheck Protection Program Flexibility Act of 2020 on June 22, 2020. The proceeds were recorded as debt, bear interest at 1% per annum and were unsecured. Amounts received under the PPP were used entirely to fund payroll costs as defined in the CARES Act and are expected to be eligible for forgiveness. As of December 31, 2021, the Company had not met the criteria for loan forgiveness. As such, the $503,950 of PPP funding is presented as long-term debt as of December 31, 2021.
On April 4, 2022, the Company received notification the PPP Loan amount of $503,950 had been fully forgiven by the SBA. Accordingly, forgiveness of the PPP loan was included in other income for the six months ended June 30, 2022.
Based on current SBA guidance, the SBA has 6 years (up to 2026) to audit the good faith certification of eligibility and expenditures related to the Company’s PPP loan proceeds.
Note 10 - Related Party Transactions
The majority stockholder of the Company is also an employee of the Company. Salaries paid to the majority stockholder for the six months ended June 30, 2023 and 2022 were $225,000 and $322,500, respectively, and are included in selling, general, and administrative expenses in the accompanying statement of operations. The Company made distributions of $0 and $12,160 for the six months ended June 30, 2023 and 2022, respectively to the majority stockholder.
| F-60 |
VILLAGE OAKS PATHOLOGY SERVICES, P.A.
D/B/A PRECISION PATHOLOGY SERVICES
NOTES TO UNAUDITED FINANCIAL STATEMENTS
Note 11 - Commitments and Contingencies
Litigation
From time to time, the Company may become subject to legal proceedings, claims or litigation arising in the ordinary course of business. In addition, the Company may receive notices alleging infringement of patents or other intellectual property rights. If an unfavorable outcome were to occur in litigation, the impact could be material to the Company’s business, financial condition, cash flow or results of operations, depending on the specific circumstances of the outcome. The Company accrues for loss contingencies when it is both probable that the Company will incur the loss and when it can reasonably estimate the amount of the loss or range of loss. As of June 30, 2023 and December 31, 2022, no amounts were required to be accrued for loss contingencies.
bioAffinity Technologies, Inc. License Agreement
The Company has a license with bioAffinity Technologies, Inc. (“bioAffinity”) which allows the Company the use of bioAffinity’s proprietary CyPath® technology to provide patients with a diagnostic test for the detection of cancer. The license has an initial term through the date that the Company obtains FDA approval to directly commercialize similar equipment (or a functional equivalent of the licensed equipment). This license provides for certain royalties based on a percentage of services rendered. As of June 30, 2023 and December 31, 2022, there have been no amounts required to be accrued for under the license agreement.
Note 12 - Retirement Plan
The Company maintains a 401(k) plan for qualified employees. The plan covers substantially all full-time employees of the Company who meet certain age and length of service requirements. There is no requirement for the Company to match employee contributions to the plan. The Company did not contribute to the plan for the periods ending June 30, 2023 and 2022.
Note 13 - Subsequent Events
The Company has evaluated subsequent events occurring after the balance sheet date through the date of September 19, 2023 which is the date the financial statements were available to be issued. Based on this evaluation, the Company has determined the following subsequent events have occurred which require adjustment disclosure in the financial statements.
On September 18, 2023, the Company entered into an Asset Purchase Agreement (the “Asset Purchase Agreement”) wherein the Company was acquired by Precision Pathology Laboratory Services, LLC, a Texas limited liability company (“PPLS”), that is a wholly owned subsidiary of bioAffinity Technologies, Inc. (“bioAffinity”). Pursuant to the terms of the Asset Purchase Agreement the Company received $3,500,000 in consideration for the assets to be purchased by PPLS, of which $1,000,000 was paid by the issuance of 564,972 shares of bioAffinity’s restricted Common Stock to a trust controlled by Dr. Joyce (the “Joyce Trust”), which share number was based on the average of the trading day closing prices of bioAffinity for the 30 days prior to September 15, 2023, rounded to the nearest whole share.
| F-61 |
PRELIMINARY PROSPECTUS
bioAffinity Technologies, Inc.
![]()
3,086,419 Units
Each Unit Consisting of
One Share of Common Stock,
One Warrant to Purchase One Share of Common Stock,
(and the shares of Common Stock underlying such Warrant)
WallachBeth Capital, LLC
September [●], 2023
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
Item 13. Other Expenses of Issuance and Distribution
The following table sets forth all expenses to be paid by the registrant, other than estimated underwriting discounts and commissions, in connection with this Offering. All amounts shown are estimates except for the Securities and Exchange Commission registration fee, the Financial Industry Regulatory Authority (FINRA) filing fee and the exchange listing fee:
| Amount to be Paid | ||||
| Securities and Exchange Commission registration fee | $ | 1,500 | ||
| FINRA filing fee | 2,500 | |||
| Printing and engraving expenses | 5,995 | |||
| Legal fees and expenses | 250,000 | |||
| Accounting fees and expenses | 60,000 | |||
| Transfer agent and registrar fees | 598 | |||
| Miscellaneous | 240,000 | |||
| Total | $ | 560,593 | ||
Item 14. Indemnification of Directors and Officers
bioAffinity Technologies, Inc. is incorporated under the laws of the State of Delaware. Reference is made to Section 102(b)(7) of the DGCL, which enables a corporation in its original certificate of incorporation or an amendment thereto to eliminate or limit the personal liability of a director for violations of the director’s fiduciary duty, except (1) for any breach of the director’s duty of loyalty to the corporation or its stockholders, (2) for acts or omissions not in good faith or which involve intentional misconduct or a knowing violation of law, (3) pursuant to Section 174 of the DGCL, which provides for liability of directors for unlawful payments of dividends or unlawful stock purchase or redemptions, or (4) for any transaction from which the director derived an improper personal benefit.
Section 145(a) of the DGCL provides, in general, that a corporation may indemnify any person who was or is a party or is threatened to be made a party to any threatened, pending or completed action, suit or proceeding, whether civil, criminal, administrative or investigative (other than an action by or in the right of the corporation), because such person is or was a director, officer, employee or agent of the corporation, or is or was serving at the request of the corporation as a director, officer, employee or agent of another corporation, partnership, joint venture, trust or other enterprise, against expenses (including attorneys’ fees), judgments, fines and amounts paid in settlement actually and reasonably incurred by the person in connection with such action, suit or proceeding, if he or she acted in good faith and in a manner he or she reasonably believed to be in or not opposed to the best interests of the corporation and, with respect to any criminal action or proceeding, had no reasonable cause to believe his or her conduct was unlawful.
Section 145(b) of the DGCL provides, in general, that a corporation may indemnify any person who was or is a party or is threatened to be made a party to any threatened, pending or completed action or suit by or in the right of the corporation to procure a judgment in its favor because the person is or was a director, officer, employee or agent of the corporation, or is or was serving at the request of the corporation as a director, officer, employee or agent of another corporation, partnership, joint venture, trust or other enterprise, against expenses (including attorneys’ fees) actually and reasonably incurred by the person in connection with the defense or settlement of such action or suit if he or she acted in good faith and in a manner he or she reasonably believed to be in or not opposed to the best interests of the corporation, except that no indemnification shall be made with respect to any claim, issue or matter as to which he or she shall have been adjudged to be liable to the corporation unless and only to the extent that the adjudicating court determines that, despite the adjudication of liability but in view of all of the circumstances of the case, he or she is fairly and reasonably entitled to indemnity for such expenses which the adjudicating court shall deem proper.
Section 145(g) of the DGCL provides, in general, that a corporation may purchase and maintain insurance on behalf of any person who is or was a director, officer, employee or agent of the corporation, or is or was serving at the request of the corporation as a director, officer, employee or agent of another corporation, partnership, joint venture, trust or other enterprise against any liability asserted against such person and incurred by such person in any such capacity, or arising out of his or her status as such, whether the corporation would have the power to indemnify the person against such liability under Section 145 of the DGCL.
In addition, as permitted by Delaware law, our Charter includes provisions that eliminate the personal liability of our directors for monetary damages resulting from breaches of certain fiduciary duties as a director, except to the extent such an exemption from liability thereof is not permitted under the DGCL. The effect of these provisions is to restrict our rights and the rights of our stockholders in derivative suits to recover monetary damages against a director for breach of fiduciary duties as a director, subject to certain exceptions in which case the director would be personally liable. If Delaware law is amended to authorize corporate action further eliminating or limiting the personal liability of directors, then the liability of our directors will be eliminated or limited to the fullest extent permitted by Delaware law, as so amended. Our Charter does not eliminate the duty of care owed by our directors and officers and, in appropriate circumstances, equitable remedies, such as injunctive or other forms of non-monetary relief, remain available under Delaware law. This provision also does not affect the responsibilities of directors and officers under any other laws, such as the federal securities laws or other state or federal laws.
Our Charter also provides that any amendment, repeal or modification of such article unless otherwise required by law will not adversely affect any right or protection existing at the time of such repeal or modification with respect to any acts or omissions occurring before such repeal or amendment of a director serving at the time of such repeal or modification.
Our Charter and A&R Bylaws provides that we shall indemnify each of our directors, officers, employees and agents, to the fullest extent permitted by the DGCL as the same may be amended (except that in the case of an amendment, only to the extent that the amendment permits us to provide broader indemnification rights than the DGCL permitted us to provide prior to such the amendment) against any and all liability and loss suffered and expenses (including attorneys’ fees) reasonably incurred by the director, officer or such employee or on the director’s, officer’s or employee’s behalf in connection with any threatened, pending or completed proceeding or any claim, issue or matter therein, to which he or she is or is threatened to be made a party because he or she is or was serving as a director, officer or employee of our Company, or at our request as a director, partner, trustee, officer, employee or agent of another corporation, partnership, joint venture, trust, employee benefit plan or other enterprise, if he or she acted in good faith and in a manner he or she reasonably believed to be in or not opposed to the best interests of our Company and, with respect to any criminal proceeding, had no reasonable cause to believe his or her conduct was unlawful. The Charter and A&R Bylaws further provide for the advancement of expenses.
| II-1 |
In addition, the A&R Bylaws provide that the right to indemnification and advancement of expenses shall not be exclusive of any other right now possessed or hereafter acquired under any statute, provision of the Charter or A&R Bylaws, agreement, vote of stockholders or otherwise. Furthermore, our A&R Bylaws authorize us to provide insurance for our directors, officers, employees and agents against any liability, whether we would have the power to indemnify such person against such liability under the DGCL or the A&R Bylaws.
We also maintain a general liability insurance policy which covers certain liabilities of directors and officers of our Company arising out of claims based on acts or omissions in their capacities as directors or officers.
In any underwriting agreement we will enter into in connection with the sale of the Units being registered hereby, the underwriters will agree to indemnify, under certain conditions, us, our directors, our officers and persons who control us within the meaning of the Securities Act, against certain liabilities.
Item 15. Recent Sales of Unregistered Securities
The Company has not issued unregistered securities to any person within the last three years, except as described below. None of these transactions involved any underwriters, underwriting discounts or commissions, except as specified below, or any public offering, and, unless otherwise indicated below, the Company believes that each transaction was exempt from the registration requirements of the Securities Act by virtue of Section 4(a)(2) thereof, Rule 701 of the Securities Act and/or Rule 506 of Regulation D promulgated thereunder. All recipients had adequate access, though their relationships with the Company, to information about the Company.
Between January 14, 2019 and July 27, 2020, we issued and sold secured, convertible promissory notes to 25 investors pursuant to a note purchase agreement with an aggregate principal amount of $3,451,326. These notes bear interest at 8% per annum and originally had a maturity date of May 31, 2022. Except as to one note with a principal amount of $100,000 that has been repaid in full, these notes have been amended and currently have a maturity date of October 31, 2022. The principal and accrued interest under these notes will automatically convert into shares of the equity security sold by the Company in its next equity financing involving the receipt by the Company of at least $5,000,000 at a price per share equal to 80% of the lowest per share price paid for such securities by the investors in such offering. Holders of these notes may also, at their option, convert the principal and accrued interest under their notes (or any portion thereof) into shares of the Company’s Common Stock at a price per share equal to $4.20 per share.
Between October 22, 2020 and June 8, 2021, we issued and sold unsecured, convertible promissory notes to 10 investors with an aggregate principal amount of $937,957. These notes bear interest at 8% per annum and had a maturity date of May 31, 2022. These notes have been amended and currently have a maturity date of October 31, 2022. The principal and accrued interest under these notes will automatically convert into shares of the equity security sold by the Company in its next equity financing involving the receipt by the Company of at least $5,000,000 at a price per share equal to 80% of the lowest per share price paid for such securities by the investors in such offering. Holders of these notes may also, at their option, convert the principal and accrued interest under their notes (or any portion thereof) into shares of the Company’s Common Stock at a price per share equal to $4.20 per share.
Between June 30, 2021 and August 28, 2021, we issued and sold unsecured, convertible promissory notes to 6 investors with an aggregate principal amount of $870,000. These notes bear interest at 8% per annum and have a maturity date of December 31, 2022. The principal and accrued interest under these notes automatically converted into shares of the Company’s Common Stock upon completion of our initial public offering at the initial public offering price of $6.125. Holders of these notes also, at their option, prior to our initial public offering, could have converted the principal and accrued interest under their notes (or any portion thereof) into shares of the Company’s Common Stock at a price per share equal to $4.20 per share.
Between October 7, 2021 and January 20, 2022, we issued and sold unsecured, convertible promissory notes to 23 investors pursuant to a note purchase agreement with an aggregate principal amount of $2,425,000. These notes bear interest at 6% per annum and had a maturity date of May 31, 2022. These notes have been amended and currently have a maturity date of October 31, 2022. The principal and accrued interest under these notes automatically converted into shares of the Company’s Common Stock upon completion of our initial public offering at the initial public offering price of $6.125. Holders of these notes also, at their option, prior to our initial public offering, could have converted the principal and accrued interest under their notes (or any portion thereof) into shares of the Company’s Common Stock at a price per share equal to $4.20 per share. Pursuant to the terms of the note purchase agreement, each of these notes was accompanied by warrants to purchase that number of shares of the Company’s Common Stock equal to the principal amount of the note divided by $4.20. Accordingly, warrants to purchase up to 577,380 shares of the Company’s Common Stock were issued to the noteholders. These warrants have an exercise price of $5.25 per share because our initial public offering was not completed on or before May 31, 2022. The warrants have a term of 5 years. In addition, we issued a warrant to the placement agent in the convertible note offering exercisable for 29,464 shares of our Common Stock at an exercise price equal to 120% of the initial public offering price of $6.125.
Between November and December of 2021, we issued warrants to the holders of our convertible promissory notes issued prior to June 30, 2021 as consideration for their agreement to extend the maturity date of such notes to May 31, 2022. We issued warrants to purchase that number of shares of Common Stock equal to the original principal amount of the notes divided by $4.20. Accordingly, we issued warrants to purchase 1,419,509 shares of the Company’s Common Stock to these noteholders. These warrants have an exercise price of $5.25 per share because our initial public offering was not completed on or before May 31, 2022. The warrants have a term of five years.
In July of 2022, we issued 729,658 warrants to the holders of our convertible promissory notes that agreed to an extension of the maturity date of such notes to October 31, 2022. The warrants are exercisable to purchase Common Stock at an exercise price of $5.25 per share. The warrants have a term of five years.
| II-2 |
In connection with the sale of the convertible bridge notes and issuance of the warrants in the fourth quarter of 2021 and the first quarter of 2022 (none of which were purchased by the Placement Agent), we have agreed to issue to WallachBeth Capital, LLC, the exclusive placement agent for the convertible bridge notes and the associated warrants, the Placement Agent’s Warrant to purchase one share of Common Stock based on the investors’ bridge note principal balance investment, or a total of 29,464 shares of our Common Stock. The exercise price of one share of our Common Stock pursuant to the Placement Agent’s Warrant is $7.35 (120% of the initial public offering price of one Unit). The Placement Agent’s Warrant is non-exercisable for 180 days following the commencement of sales in our initial public offering and will expire on a date that is not more than five (5) years from the date of the commencement of sales in our initial public offering in compliance with FINRA Rule 5110(e)(1)(A). The Placement Agent’s Warrant has been deemed compensation by FINRA and is therefore subject to a 180-day lock-up pursuant to FINRA Rule 5110(e)(1). The Placement Agent (or its respective permitted assignees under Rule 5110(e)(2)(B)) will not sell, transfer, assign, pledge, or hypothecate the Placement Agent’s Warrant or the securities underlying such warrant, nor will they engage in any hedging, short sale, derivative, put, or call transaction that would result in the effective economic disposition of such warrant or the underlying securities for a period of 180 days following the date of commencement of sales in our initial public offering. The Placement Agent’s Warrant contains the same adjustment provisions as the warrants issued to the investors in the bridge notes. In addition, we have granted the underwriters the ability to exercise them in a “cashless” manner. The Placement Agent’s Warrant and the underlying shares of Common Stock that may be issued upon exercise have been registered.
Between April 2014 and March 2022, we issued non-statutory stock options under our 2014 Stock Incentive Plan to certain of our employees, directors and consultants to purchase up to 969,645 shares of our Common Stock. Some of those options were exercised, resulting in the issuance of 34,456 shares of our Common Stock. Options to purchase 55,380 shares of our Common Stock were forfeited when the recipients’ service to the Company was terminated. Options to purchase 876,952 shares of our Common Stock at a weighted average exercise price of approximately $4.13 per share remain outstanding as of the date of this registration statement. The options generally have a term of 10 years from the date of grant.
Between August 2015 and January 2022, we issued 41,417 shares of our Common Stock as restricted stock grants under our 2014 Stock Incentive Plan to certain of our employees and consultants.
In August of 2022, we issued and sold unsecured, convertible promissory notes to two investors pursuant to a note purchase agreement with an aggregate principal amount of $249,000. These notes bear interest at 6% per annum and have a maturity date of October 31, 2022. The principal and accrued interest under these notes automatically converted into shares of the Company’s Common Stock upon completion of our initial public offering at the initial public offering price of $6.125. Holders of these notes also, at their option, prior to our initial public offering, could have converted the principal and accrued interest under their notes (or any portion thereof) into shares of the Company’s Common Stock at a price per share equal to $4.20 per share. Pursuant to the terms of the note purchase agreement, each of these notes was accompanied by warrants to purchase that number of shares of the Company’s Common Stock equal to the principal amount of the note divided by $4.20. Accordingly, warrants to purchase up to 59,285 shares of the Company’s Common Stock were issued to the noteholders. These warrants have an exercise price equal to $5.25 per share. The warrants have a term of 5 years.
On January 1, 2023, we issued an aggregate of 57,589 restricted shares of the Company’s Common Stock to our seven directors, which shares of restricted stock will vest ratably over three months of continued service and which represents a restricted stock award to each director valued at $18,750 granted by us to each of our directors each quarter during the calendar year as part of our director compensation policy.
On April 15, 2023, we issued an aggregate of 69,440 restricted shares of the Company’s Common Stock to our seven directors, which shares of restricted stock will vest one-third on the date of grant, one-third on May 1, 2023 and the remaining shares on June 1, 2023, provided each individual continues to service as a director, and which represents a restricted stock award to each director valued at $18,750 granted by us to each of our directors each quarter during the calendar year as part of our director compensation policy.
On June 6, 2023, we issued 26,178 restricted shares of the Company’s Common Stock to our Chief Executive Officer and 52,356 restricted shares of the Company’s Common Stock to our Chief Financial Officer in consideration of services provided.
On July 1, 2023, we issued an aggregate of 71,715 restricted shares of the Company’s Common Stock to our seven directors, which shares of restricted stock will vest ratably over three months of continued service and which represents a restricted stock award to each director valued at $18,750 granted by us to each of our directors each quarter during the calendar year as part of our director compensation policy.
Between April 1, 2023 and September 1, 2023, we issued an aggregate of 16,048 shares of the Company’s Common Stock to a consultant pursuant to the terms of a consulting agreement in consideration of services provided.
On August 9, 2023, we issued 26,315 restricted shares of the Company’s Common Stock to an officer pursuant to the terms of his employment agreement.
On September 18, 2023, we issued 564,972 shares of the Company’s Common Stock to the Joyce Trust pursuant to the terms of the Asset Purchase Agreement.
Item 16. Exhibits and Financial Statement Schedules
(a) Exhibits
See the Exhibit Index immediately preceding the signature page hereto for a list of exhibits filed as part of this registration statement on Form S-1, which Exhibit Index is incorporated herein by reference.
(b) Financial Statement Schedules
Schedules not listed have been omitted because the information required to be set forth therein is not applicable, not material or is shown in the financial statements or notes thereto.
Item 17. Undertakings
| (a) | The undersigned registrant hereby undertakes: |
| (1) | To file, during any period in which offers or sales are being made, a post-effective amendment to this registration statement: |
| (i) | To include any prospectus required by Section 10(a)(3) of the Securities Act of 1933, as amended (the “Securities Act”); |
| (ii) | To reflect in the prospectus any facts or events arising after the effective date of the registration statement (or the most recent post-effective amendment thereof) which, individually or in the aggregate, represent a fundamental change in the information set forth in the registration statement. Notwithstanding the foregoing, any increase or decrease in volume of securities offered (if the total dollar value of securities offered would not exceed that which was registered) and any deviation from the low or high end of the estimated maximum offering range may be reflected in the form of prospectus filed with the SEC pursuant to Rule 424(b) if, in the aggregate, the changes in volume and price represent no more than a 20% change in the maximum aggregate offering price set forth in the “Calculation of Registration Fee” table in the effective registration statement; and | |
| (iii) | To include any material information with respect to the plan of distribution not previously disclosed in the registration statement or any material change to such information in the registration statement. |
| II-3 |
provided, however, that paragraphs (i), (ii) and (iii) above do not apply if the information required to be included in a post-effective amendment by those paragraphs is contained in periodic reports filed with or furnished to the SEC by the registrant pursuant to Section 13 or Section 15(d) of the Securities Exchange Act of 1934, as amended (the “Exchange Act”) that are incorporated by reference in the registration statement, or is contained in a form of prospectus filed pursuant to Rule 424(b) that is a part of the registration statement;
| (2) | That, for the purpose of determining any liability under the Securities Act, each such post-effective amendment shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof. |
| (3) | To remove from registration by means of a post-effective amendment any of the securities being registered which remain unsold at the termination of the offering. |
| (4) | That, for the purpose of determining liability under the Securities Act to any purchaser: |
| (i) | Each prospectus filed by the registrant pursuant to Rule 424(b)(3) shall be deemed to be part of the registration statement as of the date the filed prospectus was deemed part of and included in the registration statement; and |
| (ii) | Each prospectus required to be filed pursuant to Rule 424(b)(2), (b)(5), or (b)(7) as part of a registration statement in reliance on Rule 430B relating to an offering made pursuant to Rule 415(a)(1)(i), (vii), or (x) for the purpose of providing the information required by Section 10(a) of the Securities Act shall be deemed to be part of and included in the registration statement as of the earlier of the date such form of prospectus is first used after effectiveness or the date of the first contract of sale of securities in the offering described in the prospectus. As provided in Rule 430B, for liability purposes of the issuer and any person that is at that date an underwriter, such date shall be deemed to be a new effective date of the registration statement relating to the securities in the registration statement to which that prospectus relates, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof. Provided, however, that no statement made in a registration statement or prospectus that is part of the registration statement or made in a document incorporated or deemed incorporated by reference into the registration statement or prospectus that is part of the registration statement will, as to a purchaser with a time of contract of sale prior to such effective date, supersede or modify any statement that was made in the registration statement or prospectus that was part of the registration statement or made in any such document immediately prior to such effective date. |
| (5) | That, for the purpose of determining liability of the registrant under the Securities Act to any purchaser in the initial distribution of the securities, the undersigned registrant undertakes that in a primary offering of securities of the undersigned registrant pursuant to this registration statement, regardless of the underwriting method used to sell the securities to the purchaser, if the securities are offered or sold to such purchaser by means of any of the following communications, the undersigned registrant will be a seller to the purchaser and will be considered to offer or sell such securities to such purchaser: |
| (i) | Any preliminary prospectus or prospectus of the undersigned registrant relating to the offering required to be filed pursuant to Rule 424; |
| (ii) | Any free writing prospectus relating to the offering prepared by or on behalf of the undersigned registrant or used or referred to by the undersigned registrant; |
| (iii) | The portion of any other free writing prospectus relating to the offering containing material information about the undersigned registrant or its securities provided by or on behalf of the undersigned registrant; and |
| (iv) | Any other communication that is an offer in the offering made by the undersigned registrant to the purchaser. |
| (6) | That, for purposes of determining any liability under the Securities Act, each filing of the registrant’s annual report pursuant to Section 13(a) or Section 15(d) of the Exchange Act (and, where applicable, each filing of an employee benefit plan’s annual report pursuant to Section 15(d) of the Exchange Act) that is incorporated by reference in the registration statement shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof. |
| (7) | Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers and controlling persons of the registrant pursuant to the foregoing provisions or otherwise, the registrant has been advised that in the opinion of the SEC such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Securities Act and will be governed by the final adjudication of such issue. |
| (8) | The undersigned registrant hereby undertakes that: |
| (i) | For purposes of determining any liability under the Securities Act, the information omitted from the form of prospectus filed as part of this registration statement in reliance upon Rule 430A and contained in a form of prospectus filed by the registrant pursuant to Rule 424(b)(1) or (4) or 497(h) under the Securities Act shall be deemed to be part of this registration statement as of the time it was declared effective. |
| (ii) | For the purpose of determining any liability under the Securities Act, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of these securities at that time shall be deemed to be the initial bona fide offering. |
| (9) | The undersigned registrant hereby undertakes to deliver or cause to be delivered with the prospectus, to each person to whom the prospectus is sent or given, the latest annual report to security holders that is incorporated by reference in the prospectus and furnished pursuant to and meeting the requirements of Rule 14a-3 or Rule 14c-3 under the Securities Exchange Act of 1934; and, where interim financial information required to be presented by Article 3 of Regulation S-X are not set forth in the prospectus, to deliver, or cause to be delivered to each person to whom the prospectus is sent or given, the latest quarterly report that is specifically incorporated by reference in the prospectus to provide such interim financial information. |
| II-4 |
EXHIBIT INDEX
| II-5 |
| * | Filed herewith. |
| ^ | To be filed by amendment |
| + | Indicates management contract or compensatory plan. |
| II-6 |
SIGNATURES
Pursuant to the requirements of the Securities Act of 1933, the registrant has duly caused this Registration Statement to be signed on its behalf by the undersigned, thereunto duly authorized, in the city of San Antonio, Texas, on September 20, 2023.
| bioAffinity Technologies, Inc. | ||
| By: | /s/ Maria Zannes | |
| Maria Zannes | ||
| Chief Executive Officer, President, Founder, and Director | ||
POWER OF ATTORNEY
KNOW ALL BY THESE PRESENTS that each individual whose signature appears below constitutes and appoints Maria Zannes and Michael Dougherty our true and lawful attorneys and agents with full power of substitution and resubstitution, with full power to sign for us, and in our names in the capacities indicated below, any and all amendments to this registration statement, any subsequent registration statements pursuant to Rule 462 of the Securities Act of 1933, as amended, and to file the same, with all exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorney-in-fact and agent, full power and authority to do and perform each and every act and thing requisite and necessary to be done in and about the premises, as fully to all intents and purposes as he might or could do in person, hereby ratifying and confirming all that said attorney-in-fact and agent, or his substitute or substitutes, may lawfully do or cause to be done by virtue hereof. This power of attorney may be executed in counterparts.
Pursuant to the requirements of the Securities Act of 1933, this Registration Statement has been signed by the following persons in the capacities and on the dates indicated:
| Signature | Title | Date | ||
| /s/ Maria Zannes | Founder, President, Chief Executive Officer, and Director (Principal Executive Officer) | September 20, 2023 | ||
| Maria Zannes | ||||
| /s/ Michael Dougherty | Chief Financial Officer | September 20, 2023 | ||
| Michael Dougherty | ||||
| /s/ Steven Girgenti | Founder, Executive Chairman, and Director | September 20, 2023 | ||
| Steven Girgenti | ||||
| /s/ Robert Anderson | Director | September 20, 2023 | ||
| Robert Anderson | ||||
| /s/ Stuart Diamond | Director | September 20, 2023 | ||
| Stuart Diamond | ||||
| /s/ Peter S. Knight | Director | September 20, 2023 | ||
| Peter S. Knight | ||||
| /s/ Mohsin Meghji | Director | September 20, 2023 | ||
| Mohsin Meghji | ||||
| /s/ Gary Rubin | Director | September 20, 2023 | ||
| Gary Rubin | ||||
| /s/ Roby Joyce | Director | September 20, 2023 | ||
| Roby Joyce |
| II-7 |